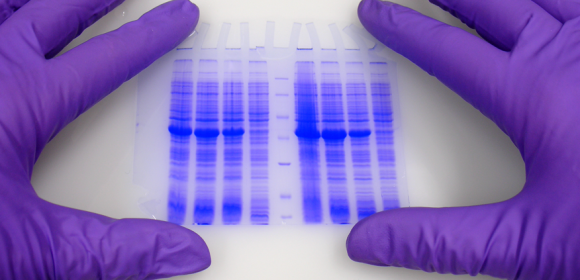

Nelle scienze della vita lo studio delle proteine è uno dei campi più importanti.
L’espressione di ciò che è codificato nei nostri geni può essere visto e, al giorno d'oggi, anche raccolto sotto il termine di Proteomica. Negli ultimi anni e decenni sono state sviluppate molte tecniche diverse. Una delle più note e utilizzate è l’elettroforesi su gel di poliacrilammide (PAGE).
Polyacrylamide Gel Electrophoresis, PAGE
Polyacrylamide Gel Electrophoresis is typically used for separation of proteins, but also DNA (especially to visualize differences between small fragments) can be separated by PAGE. The matrix consists of acrylamide-strands cross-linked wit N,N-methylenebisacrylamide. ITW Reagents offers reagents and ready mixes for SDS-PAGE, native PAGE, denaturing and non-denaturing DNA-PAGE as well as for sequencing gels.
Acrylamide mixes
The separation capacity of a polyacrylamide gel is determined by the mixing ratio of acrylamide to bisacrylamide. The lower the ratio of these two components in the mixture, the higher the degree of crosslinking. This means that a 6 % gel prepared from a stock solution of a mixing ratio of 29 : 1, has a higher degree of crosslinking than a 6 % gel prepared from a stock solution of a mixing ratio of 37.5 : 1.
For most applications an acrylamide : bisacrylamide ratio of 29 : 1 or 37.5 : 1 is used (for electrophoretic separation of nucleic acids or proteins). The mixing ratio of 19: 1 is the solution of choice for DNA sequencing. The preparation is simplified by using 30% or 40% aqueous acrylamide stock solutions with the desired ratio.
Acrylamide solutions are often termed as "gas-stabilized", offering increased shelf-life. The "gas" simply is oxygen, which is meant to avoid a spontaneous polymerization of acrylamide solutions. Since the polymerization initiators APS and TEMED are added in excess, however, "degassing" (partly done in laboratory prior to the polymerization initiation) is actually unnecessary.
The unwanted spontaneous polymerization is started by radicals which typically originate from acrylic acid. Poorer qualities often contain detectable traces of acrylic acid and solutions based on these grades bear a high risk to polymerize spontaneously. In contrast, grades recrystallized four times ("4K"), however, are free of acrylic acid. For our Molecular biology grade only 4K acrylamide especially tested for the absence of DNases, RNases and proteases, is employed!
At low temperatures of 2-8°C, the oxygen exchange is reduced within the acrylamide solution and spontaneous polymerization is facilitated. Therefore, we recommend storage at ambient temperature. The following table lists the most common acrylamide mixes; more acrylamide solutions are available or on request.
Usage of acrylamide mixes
The acrylamide stock solutions are diluted in order to obtain the desired monomer concentration. The total volume of the required amount of gel solution (for example, 100 ml) is divided by the content of the solution (e.g. 30 %) and multiplied with the desired final concentration (for example 6 % acrylamide) to obtain the required volume of the stock solution:
(100 ml / 30 %) * 6 % = 20 ml
To prepare 100 ml of a 6 % acrylamide gel, 20 ml of the 30 % acrylamide stock solution is added into the gel-mixture.
Of course, you can request our acrylamide mixes in powder form as well. But caution, the acrylamide monomer is a strong accumulating neurotoxin! Therefore, gloves and a face mask should be worn when handling crystalline acrylamide.

Buffer solutions, SDS, APS, TEMED
The most commonly used electrophoresis buffer for SDS-PAGE is SDS-tris-glycine, the so-called Laemmli buffer. An alternative is the tris-tricine-SDS system invented by Schaegger & Jagow. For native protein gels, tris-glycine buffer is the first choice.
To initiate the polymerization, 100 µl of 10 % APS and 5-10 µl TEMED per 10 ml gel solution are added. Since the polymerization is very fast induced by TEMED and the radical initiator APS, the gel should be poured immediately. Cooling down the solution decelerates the polymerization process. APS (ammonium persulfate, A1142) is not very stable in aqueous solution (commonly a 10 % stock solution in water is prepared), but from experience, the solution can be stored at 2-8°C for several weeks, or at -20°C for months without losing its activity.
TEMED (tetramethylethylenediamine, A1148) enhances the polymerization of acrylamide and bisacrylamide by catalysing the formation of free radicals of APS. It is used in a concentration of 50 µl / 100 ml of gel solution. The stability of TEMED is very high (2 years), if contact to water is prevented.
Buffer components and other chemicals for Polyacrylamide Gel Electrophoresis of proteins
| Prod. No. | Description | Comment |
|---|---|---|
| A1142 | Ammonium persulfate | APS; the recommended final concentration in acrylamide gels is 0.1 % (w/v). Prepare a 10 % (w/v) stock solution for an easy application. |
| A1148 | TEMED | N,N,N',N'-Tetramethylethylenediamine; the recommended final concentration in acrylamide gels is 0.1 % (v/v). |
| A0676 | SDS-Solution 10 % | Sodium dodecylsulfate; also available as 20 % solution (A0675) or crystalline (e.g. A2572). |
Ready-to-use Acrylamide solutions for SDS-PAGE
Save time and assure reproducible results: Our ready-to-use gel solutions for SDS-PAGE are your product of choice for an easy and fast gel preparation. ITW Reagents offers ready-to-use solutions for stacking and resolving gels according to Laemmli, based on an acrylamide/bisacrylamide ratio of 29 : 1 with 0.1% SDS and Tris.
To prepare the acrylamide gel, 100 ml of each ready-to-use solution (see table below) is completed with 1 ml APS (from a 10% stock solution based on A1142) and 50 µl TEMED (A1148) to initiate the polymerization of the gel. For electrophoresis, a SDS-tris-glycine buffer (for example, A1415) is used.
Protein markers and protein dyes
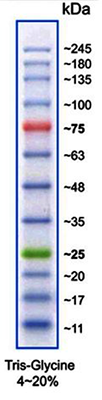
(Image: Protein Marker VI)
Protein size standards for SDS-PAGE
| Prod. No. | Description | Comment |
| A8889 | Protein Marker VI (10-245) prestained | 12 bands; ready-to-use and prestained marker for better detection and orientation |
For non-specific staining of the protein bands on the polyacrylamide gel mainly Coomassie® Brilliant Blue R-250 is used. If higher sensitivity is required, silver nitrate staining is employed. Before or during the staining procedure, the proteins need to be fixed in the gel matrix. This is done by an aqueous mixture of ethanol and acetic acid or trichloroacetic acid that lead to protein denaturation and precipitation. For fixing of small basic proteins, formaldehyde is more suitable.
Protein dyes for staining of polyacrylamide gels
| Prod. No. | Description | Comment |
| A1092 | Coomassie® Brilliant Blue R-250 | One of the most commonly used stains for proteins in SDS-PAGE. The protein-dye complex has an absorption maximum at 549 nm. Per positively charged amino acid approximately 1.5 - 3 molecules of Coomassie® Brilliant Blue R-250 will be bound. The sensitivity is around 200-400 ng protein/band. |
| A3480 | Coomassie® Brilliant Blue G-250 | The main application of this dye is the Bradford assay (determination of protein concentration), but staining of proteins in polyacrylamide gels is possible as well. |
| A3930 | Rhodamine B  |
See Eriochrome black T (131439). |

The history of electrophoresis - past and present
Charged molecules migrate in an electric field. This is the basic principle of electrophoresis, a method used in almost every biology lab today and fundamental for most separation techniques and analytic methods.
When we hear the term electrophoresis we will probably think of gel electrophoresis, agarose or acrylamide, especially SDS-PAGE – which is today the most popular and most widely used electrophoretic method in research worldwide. But the field of electrophoretic methods and applications is very broad and, in contrast to the now dominating techniques, the first steps of electrophoresis were performed in free solution without any kind of support medium.
I primi anni
Com'è iniziata questa storia di successo? La teoria di base dell'elettroforesi fu sviluppata più di 200 anni fa, ma solo nel 1930 Tiselius presentò la cosiddetta elettroforesi a fronte mobile. Questa tecnica di elettroforesi a soluzione libera era adatta per studiare la mobilità delle molecole caricate. Nel “apparato di Tiselius” il “fronte mobile” formato da proteine migranti elettroforeticamente è stato misurato dalle variazioni di assorbimento della luce o dall'indice di rifrazione. Contrariamente ai metodi odierni, una separazione completa dei componenti di una miscela non fu mai raggiunta, indipendentemente dalla durata dell'esperimento. Fu possibile solo un'analisi parziale dei composti di migrazione più rapidi e più lenti.
Negli anni '50, l'elettroforesi a fronte mobile fu superata dall'elettroforesi di zona, un diverso principio di elettroforesi, descritto per la prima volta nel 1939. Nell’elettroforesi di zona, proteine, nucleosidi, amminoacidi e altre molecole erano fisicamente separate l'una dall'altra con l'aiuto di un mezzo di supporto. All'inizio sono stati utilizzate carte filtranti; ma presto il metodo fu fortemente migliorato da supporti alternativi come l'acetato di cellulosa (Kohn 1957), l'amido (Smithies 1955), il poliacrilammide (Raymond & Weintraub 1959) e l'agarosio (Hjertén 1961). Il mezzo di supporto impedisce alle molecole di sedimentare e, a differenza dell'elettroforesi a fronte mobile, consente quindi una completa separazione.
L’invenzione della PAGE
Negli anni '60, è iniziata una nuova era nel campo dell'elettroforesi. Sono state introdotte nuove tecniche e i metodi esistenti sono stati in gran parte migliorati. Sono state sviluppate l'elettroforesi su disco e la messa a fuoco isoelettrica. Inizialmente il blu di Coomassie® è stato utilizzato come colorante per visualizzare le bande proteiche dopo l’elettroforesi su gel e la SDS è stata identificata per "mascherare" la carica netta di proteine durante l'elettroforesi del poliacrilammide.
Cinque anni dopo che Raymond & Weintraub introdussero i gel di poliacrilammide per l'elettroforesi proteica, questo materiale fu usato da Ornstein e Davis come supporto per una tecnica che è nota e applicata in quasi tutti i laboratori biochimici fino ad oggi: Nel 1964, hanno sviluppato l'elettroforesi a disco (discontinua), che combina l'elettroforesi con l’isotacoforesi.
Isotachophoresis is performed in a discontinuous buffer system
By isotachophoresis - initially termed as ion migration method (Kendall & Crittenden 1923) or displacement electrophoresis (Martin 1942) - sharp boundaries between the sample constituents can be generated. Unlike zone electrophoresis, isotachophoresis is performed in a discontinuous buffer system, composed of a leading electrolyte of high mobility and a terminating or trailing ion of low mobility. After setting up the electric field, the molecules migrate at different speeds and therefore completely separate from each other forming stacks; the molecule with the highest mobility directly follows the leading ion, molecules with the lowest mobility migrate directly in front of the terminating electrolyte. Nothing special so far. However, the faster moving ions will lower the surrounding electrical field while the slower ones create a higher field. As a consequence, after separation, all molecules migrate with the same speed (and this is what the name isotachophoresis literally means: migration at equal speed): the initially faster migrating ions will be retarded by the surrounding weaker field; the slower moving ions will be accelerated by the stronger field around them. For this reason, the system has a self-sharpening effect; as soon as an ion diffuses out of its own band it will either be de-accelerated or accelerated and so forced backwards into its original band.
Back again to the principle of disc electrophoresis. By using 2 different areas of separation (stacking and resolving gel) and a discontinuous buffer system, Ornstein and Davis prevented the formation of protein aggregates during the entry into the gel matrix and supported the separation to well defined bands.
The stacking gel is characterized by a lower pH and larger pores than the adjacent resolving gel.
The stacking gel is characterized by a lower pH and larger pores than the adjacent resolving gel. Both gels contain only chloride ions (with Tris as a counter ion), while the electrode buffer contains only glycine. At first, the proteins are “stacked” and concentrated by the principle of isotachophoresis. Due to the large pores of the stacking gel, the size of the molecule does not influence their mobility. The net charge of glycine is almost zero at the pH of the stacking gel, therefore glycine functions as terminating ion. When the protein front reaches the border to the close-meshed resolving gel, the small glycine molecules pass through the proteins, enters the resolving area, becomes higher charged and moves together with the chloride ions in front of the protein fraction. As soon as the proteins are surrounded by the homogeneous buffer they start separating according to the principles of zone electrophoresis: now their mobility depends on their charge and size, which finally leads to a rearrangement of the protein-ranking.
When Laemmli in 1970 published his famous paper on T4 phage protein separation, he used Ornstein and Davis’ Tris-glycine-chloride buffer system. But instead of conducting a “native” PAGE, he added SDS, which was introduced by Shapiro et al. in 1967. In SDS-PAGE, the separation process is no longer based on the specific net charge of the proteins, but on their differences in molecular weight. The combination of SDS-PAGE with “Laemmli”-buffer is the most frequently used technique for separation of proteins till this date.
Another milestone in the development of gel electrophoresis was the isoelectric separation of proteins. The theory of isoelectric focusing was introduced by Svensson already in 1961, but only after Vesterberg successfully synthesized carrier ampholytes for generation of a continuous pH gradient, the technique of isoelectric focusing was born. In 1975, O’Farrell combined isoelectric focusing with SDS-PAGE; the so-called 2D-electrophoresis (or “protein-mapping”, since every protein is characterized by a specific position due to its intrinsic charge and mass) became an important tool for protein analysis.
A quantum jump in protein detection in acrylamide gels (especially 2D-electrophoresis) was the introduction of silver staining techniques by Merrill et al. 1979. Compared to the previously predominant Coomassie® staining, the sensitivity was strongly increased from micrograms to nanograms. In the same year, Towbin et al. performed the first Western Blot; he transferred SDS-PAGE-separated proteins to a nitrocellulose membrane.
SDS-PAGE: Is there anything after Laemmli?
The Laemmli-coined performance of SDS-PAGE is present until now, so we have to ask if there are no alternatives. Is this the end of evolution in SDS-PAGE? No further techniques or improvements during the last 40 years? – Sure there are some!
There are a few classes of proteins that behave anomalously in SDS-PAGE: glycoproteins, strongly basic proteins (positively charged) and some hydrophobic transmembrane proteins. Regarding the highly hydrophilic glycoproteins, the usage of alkaline Tris-borate-EDTA buffer (Poduslo 1981) provides a solution. To separate histones, Panyim & Chalkley developed acid urea polyacrylamide gels (AU gels) in 1969. An alternative is the TAU gel, which additionally contains the non-ionic detergent Triton®. TAU gels are suitable for the identification of modifications of proteins such as acetylation and phosphorylation. Other highly charged proteins can be separated according to their size by using the cationic detergent cetyltrimethylammonium bromide instead of SDS (Eley et al. 1979).
For an improved separation of small peptides Schägger & Jagow used an alternative Tris-Tricine-buffer system for SDS-PAGE in 1987. Four years later the same researchers introduced a discontinuous electrophoretic system for the isolation of membrane proteins from acrylamide gels. In their blue native electrophoresis, Coomassie instead of SDS is used to induce a charge shift on the proteins. In addition, aminocaproic acid and non-ionic detergents like Triton® X-100 serve to improve solubilization of the membrane proteins. The resulting dye-detergent-protein complexes are separated within a aminocaproic acid-containing PAG and, for studies on quaternary structure, resolved into the individual polypeptides by second-dimension Tricine-SDS-PAGE. For small membrane proteins, Coomassie® is proposed to be replaced by taurodeoxycholate.
The beauty of simplification - Ahn “Single gels”
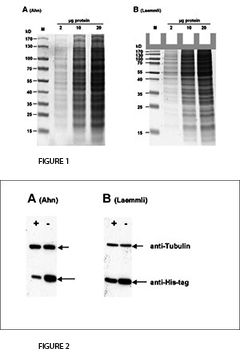
In 2001, Ahn et al. introduced a simplified variant of the SDS-PAGE procedure that consists of only one “single gel” with an increased length for the separating gel. In Laemmli’s Tris-glycine gels, glycine serves as the slow moving ion. In contrast, Ahn single gels employ three amino acids for this purpose, namely glycine, serine, and asparagine. In Laemmli’s original protocol the separating gel works at pH value 8.8, whereas Ahn’s single gel works at pH 7.4. At such a mild alkaline pH value the hydrolysis of acrylamide is minimized. It improves stability and shelf life of acrylamide solutions and gels. Ahn gels do not contain any detergent but SDS is added to the running buffer to permit denaturing conditions during electrophoresis. In the original paper of Ahn et al., no comparison to other SDS-PAGE variants is shown. We wanted to study the differences in the performance of gels according to Ahn or Laemmli. To this end we conducted gel electrophoresis on 10 % polyacrylamide gels according to both of the protocols. Additionally, we investigated the transfer of proteins onto blotting membranes in a Western blot assays. Our results in brief:
1. Electrophoresis.
The fist clear difference between Laemmli and Ahn gels is the length of the separation gel for the same type of gel cassette. Single gels according to Ahn make longer separation gels (Fig. 1, SDS-PAGE of total protein from HEK293T cell lysate using 10 % acrylamide gels according to Ahn et al. (A) and Laemmli (B). Coomassie® stained gels show about 20 prominent protein bands with different size distributions for both gel types. Migration patterns of proteins were different for both gel types. Proteins with molecular mass higher than 70 kD appear compressed on Laemmli Tris-glycine gels, whereas marker proteins were distributed linear over a wide range of sizes using an Ahn single gel.). However, the migration patterns for proteins differ between Laemmli Tris-gylcine and Ahn single gels at different molecular weight ranges. For proteins of 15 – 70 kilo Dalton (kD) 10 % Laemmli gels show the highest resolution whereas 10 % Ahn gels show a linear migration pattern for protein sizes ranging from 25 – 130 kD. When trying to separate a wide range of protein sizes on the same gel there are two options: Ahn single gels appear particularly well suited for medium and high molecular weight proteins. Laemmli Tris-glycine gels are best for small and medium sized proteins when used at the same concentration of acrylamide polymer.
2. Stability.
We also studied the stability of acrylamide solutions according to Ahn et al. by storing them for a period of up to six months at 2-8°C. Polyacrylamide gels made from such solutions did not show any significant effect on quality even after prolonged storage. However, cast gels should be used within a few days since the pH value within the gel changes rapidly. This holds true for Laemmli Tris-glycine gels as well as for Ahn single gels.
3. Western Blotting.
For the transfer of proteins the performance varies depending on the molecular mass of the proteins (Fig. 2, Western blot of total protein from HEK293T cells after SDS-PAGE according to Ahn et al. (A) and Laemmli (B). Two different proteins were detected with specific antibodies: Tubulin and a 25 kD his-tagged protein. Expression of Tubulin was the same in all cells. The cellular (or proteasomal) protein degradation of the his-tagged protein was induced (+) or not induced (-) in the experiments. Tubulin was transferred more efficiently from Ahn gels to PVDF membranes than from Laemmli Tris-glycine gels, whereas the transfer of the 25 kD his-tagged protein was less efficient from equivalent Ahn single gels.). Tubulin, a protein of 66 kD, was transferred from Laemmli gels 55 ± 4 % less efficiently compared to Ahn single gels. In contrast, the transfer of a 25 kD his-tag protein was less efficient from Ahn gels by 32 ± 12 % (n=4). Overall, the transfer of small proteins is better using Laemmli’s Tris-glycine gels, while Ahn single gels performed better for proteins of higher molecular mass.
4. Handling.
Interviewed users liked the fast and convenient procedure according to Ahn et al. The protocol is easily adopted by laboratories already using the “classical” Laemmli protocol since all apparatus, buffers and solutions are the same for both SDS-PAGE procedures. Other, more skeptical authors also evaluated Ahn’s single gel system and finally confirmed the good performance. According to G. Fritz (from the Univ. of Zürich, in Rehm 2007) additional benefits of Ahn gels are: 1. they run quite beautifully (do not tend to produce “smilies”), 2. gels work at higher polyacrylamide concentrations (up to 18 %), 3. they avoid the slimy stacking gel.
Perspectives
In research, new methods are constantly invented creating new insights. Some of the methods such as the gel electrophoresis withstand the test of time. Classical methods were improved, adjusted, and new features were added over the years. ITW Reagents is always striving to explore new or improved application protocols to share promising products with the customers.
Literature
- Ahn, T. et al. (2001) Anal. Biochem. 291, 300-303
- Davies, B.J. (1964) Ann. N. Y. Acad. Sci. 121, 404-427
- Eley et al. (1979) Anal. Biochem. 92, 411-419
- Fazekas de St. Groth, S. et al. (1963) Biochim. Biophys. Acta 71, 377-391
- Gordon, A.H. et al. (1950) Coll. Czech. Chem. Com. 15, 1
- Grabar, P. & Williams, C.A. (1953) Biochim. Biophys. Acta 10, 193-194
- Hjertén, S. (1961) Biochim. Biophys. Acta 53, 514-517
- Hjertén, S. (1967) Chromatogr. Rev. 9, 122-219
- Hjertén, S. (1983) J. Chromatogr. 270, 1-6
- Jorgenson, J.W. & Lukacs, K.D. (1981) Anal. Chem. 53, 1298
- Kendall, J. & Crittenden, F.D. (1923) Proc. Natl. Acad. Sci. USA 9, 75
- von Klobusitzky, D. & König, P. (1939) Arch. Exptl. Pathol. Pharmakol. Naunyn-Schmiedeberg 192, 271
- Kohlrausch, F. (1897) Wiedemanns Ann. (Ann. Phys. Chem.) 62, 209
- Kohn, J. (1957) Nature 180, 986
- Laemmli, U.K. (1970) Nature 227, 680-685
- Martin, A.J.P. (1942) unpublished results
- Merrill, R.C. et al. (1979) Proc. Natl. Acad. Sci. USA 76, 4335
- O’Farrell, P.H. (1975) J. Biol. Chem. 250, 4007-4021
- Ornstein, L. (1964) Ann. N. Y. Acad. Sci. 121, 321-349
- Panyim, S. & Chalkley, R. (1969) Arch. Biochem. Biophys. 130, 337-346
- Poduslo, J.F. (1981) Anal. Biochem. 114, 131
- Raymond, S. & Weintraub, L. (1959) Science 130, 711
- Reuss, F.F. (1809) Memoires de la Société Imperiale de Naturalistes de Moskou 2, 327
- Sanger, F. & Coulson, A.R. (1975) J. Mol. Biol. 94, 441-448
- Schägger, H. & von Jagow, G. (1987) Anal. Biochem. 166, 368-379
- Schägger, H. & von Jagow, G. (1991) Anal. Biochem. 199, 223-231
- Schwartz, D.C. & Cantor, C.R. (1984) Cell 37, 67-75
- Shapiro, A.L. et al. (1967) Biochim. Biophys. Res. Commun. 28, 815-820
- Smithies, O. (1955) Biochem. J. 61, 629-641
- Svensson, H. (1961) Acta Chem. Scand. 15, 325-341
- Tiselius, A. (1930) Nova Acta Regiae Societatis Scientarum Upsaliensis Ser IV, Vol. 7, No. 4
- Tiselius, A. (1937) Trans. Faraday Soc. 33, 524-531
- Towbin et al. (1979) Proc. Natl. Acad. Sci. USA 76, 4350–4354
- Vesterberg, O. (1969) Acta Chem. Scand. 23, 2653
- Weber, K & Osborn, M. (1969) J. Biol. Chem. 244, 4406
Reviews: - Everaerts, F.M., Becker, J.L., & Verheggen, T.P.E.M. (1976) “Isotachophoresis: Theory, Instrumentation, and Applications”; Elsevier, Amsterdam
- Michov, B. (1995) „Elektrophorese: Theorie und Praxis“; Walter de Gruyter & Co, Berlin
- Vesterberg, O. (1993) “A short history of electrophoretic methods”; Electrophoresis, 14, 1243-1249
- Righetti, P.G. (2005) “Electrophoresis: The march of pennies, the march of dimes”; Journal of Chromatography A, 1079, 24-40
- Westermeier, R. (2004) “Electrophoresis in Practice: A Guide to Methods and Applications of DNA and Protein Separations”; 4th ed. Wiley-VCH, Weinheim