
Im Zentrum der Molekularbiologie steht eine Spezies von Molekülen: DNA.
DNA-Moleküle werden amplifiziert und durch Transformation oder Transfektion in Organismen eingeführt, getrennt, gefärbt, mikroskopisch untersucht, manipuliert, sequenziert und so weiter.
Für alle diese Techniken ist der erste Schritt die Isolierung der DNA aus dem Ursprung von Interesse.
Diese Seite gibt Ihnen einen Überblick über die verschiedenen Methoden zur Nukleinsäureisolierung und bietet eine Reihe von Must-Have-Reagenzien, um reine DNA und/oder RNA aus verschiedenen Quellen zu erhalten.
Einleitung
Was bedeutet es technisch, wenn wir über DNA-Extraktion, Isolierung oder Reinigung sprechen - Begriffe, die oft synonym verwendet werden, um vorzugsweise reine DNA aus einer Probe zu erhalten? Welche Mechanismen stecken dahinter? Wie ist es möglich, verschiedene Arten von DNA zu trennen?
Auf dieser Seite konzentrieren wir uns auf die Isolierung der beiden DNA-Spezies, die hauptsächlich in molekularbiologischen Labors isoliert werden - genomische DNA und bakterielle Plasmide. Wir geben einen Überblick über etablierte DNA-Isolationstechniken, ihren chemischen Hintergrund und diskutieren die jeweiligen Vorteile und Grenzen.
Was das grundlegende Verfahren betrifft, so ist die DNA-Extraktion einfach und kann mit Haushaltsprodukten durchgeführt werden. Im Grunde genommen benötigen Sie nur eine reiche Quelle an DNA, Salz, Wasser, Geschirrspülmittel, einen Kaffeefilter, hochprozentigen Alkohol und einen Stick, um das gefällte DNA-Salz aus der Lösung zu spulen. Für höhere Anforderungen (in Bezug auf Quantität und Qualität) bedarf das Verfahren natürlich einer weiteren Verfeinerung.
Reinheit und Integrität der DNA beeinflussen die Ergebnisse aller nachfolgenden Anwendungen, so dass höchste Qualität der DNA für Diagnose und Forschung wünschenswert ist.
Nicht nur zu Hause, sondern auch im Labor sind die Prozessschritte der Isolation unkompliziert: Erstens: Zerstören Sie die Zellen und bringen Sie die DNA in Lösung. Zweitens: Entfernen Sie Verunreinigungen durch nichtaffinitäts- (DNA bleibt in Lösung, während alles andere entfernt wird) oder affinitäts- (DNA bindet selektiv an eine feste Matrix) basierte Techniken.
Es kann notwendig sein, Zellen zunächst aus dem Probenmaterial zu dissoziieren (z.B. bei Gewebe, Knochen und Pflanzen). Dieses Verfahren ist von Probe zu Probe unterschiedlich und verwendet fast jede physikalische Methode, die schließlich zu einer Maische aus DNA-haltigem Material führt, einschließlich Schneiden, enzymatischer Behandlung, Mischen (z.B. mit Glasperlen), Mahlen und Pulvern in einem Mörser oder Gefrieren in flüssigem Stickstoff.
Geschichte der Entdeckung der DNA
Die allererste DNA-Isolierung wurde 1869 vom Schweizer Arzt Friedrich Miescher beschrieben. Im Kern der Leukozyten, der aus dem Eiter auf chirurgischen Verbänden gewonnen wurde, identifizierte er eine säurelösliche phosphor- und stickstoffhaltige Substanz, die er " Nuklein " nannte. Für die Isolierung dieser neuen Substanz, die 1889 von seinem Schüler Richard Altmann in "Nukleinsäure" umbenannt wurde, verwendete Miescher ein aufeinanderfolgendes Lösungsmittel- und Alkaliextraktionsverfahren mit anschließender Ansäuerung. 1944, fast 50 Jahre später, begann die bekanntere Geschichte der DNA mit Avery, MacLeod und McCarty, die zeigten, dass die Erbsubstanz tatsächlich Nukleinsäuren sind (und nicht Proteine, wie in früheren Studien angenommen). Nach den Beobachtungen von Erwin Chargaff (feste Verhältnisse von Purin- und Pyrimidinbasen innerhalb einer Art) 1949 und den Röntgenuntersuchungen von Rosalind Franklin und Maurice Wilkins 1953 (Helixstruktur) entdeckten James Watson und Francis Crick schließlich die molekulare Doppelhelixstruktur der DNA, die durch die Basenpaarung von Adenin mit Thymin und Cytosin mit Guanin 1953 entstand.
Lysis
(siehe auch Enzyme für die Nukleinsäurebiochemie)
Auch wenn DNA-Moleküle buchstäblich auf der Straße zu finden sind, ist die zu untersuchende DNA typischerweise in einer Zelle eingeschlossen. Um die DNA zu erhalten, ist ein Prozess namens Lyse (griechisch λύσις, lýsis from lýein "to separate") erforderlich: Zellwand (falls vorhanden) und Membran werden abgebrochen, so dass alles, was früher in der Zelle eingeschlossen war, in Lösung freigegeben werden kann: DNA, RNA, Proteine, Lipide und Metaboliten.
Lysepuffer für genomische DNA beinhalten üblicherweise ein Detergenz, Natriumchlorid, EDTA und Enzyme zum Abbau von Proteinen und RNA.
lonische Detergenzien, wie z.B. SDS, destabilisieren stark die Lipiddoppelschicht der Zellmembran. Sie lösen die Membranlipide auf, forcieren schließlich den Abbau der Zellstruktur und unterstützen die Fällung von Lipiden und Proteinen. EDTA hilft bei der Destabilisierung der Zellwand, aber seine Hauptaufgabe ist es, die DNase-Aktivität durch Chelatbildung von zweiwertigen Kationen wie Mg2+ zu hemmen. Wenn es um den Proteinabbau geht, ist die Proteinase K die häufigste Protease, die direkt in der Lyselösung eingesetzt wird. Bedingungen, die die Enzyminaktivierung fördern, wie Detergenzien, chaotrope Salze, hohe Temperaturen und pH-Wertänderungen, werden von der Proteinase K gut vertragen. Neben ihrer hohen Stabilität zeichnet sich die Proteinase K durch eine große Anzahl von Spaltstellen aus und ist daher hervorragend geeignet, zelluläre und nukleare Proteine zu entfernen, die an die DNA gebunden sind. Auch RNA wird enzymatisch abgebaut. Die Ribonuklease RNase A, die RNA-Moleküle zu einzelnen Nukleotiden hydrolysiert, ist ebenfalls hitzebeständig und benötigt wie die Proteinase K keine Kofaktoren, die durch EDTA komplexiert werden könnten.
Im Gegensatz dazu halten andere Enzyme wie Lysozyme den Lysebedingungen nicht stand. Lysozyme verdauen effektiv die bakterielle Zellwand (bestehend aus Peptidoglykan), aber das Enzym wird durch oberflächenaktive Substanzen wie SDS gehemmt. Aus diesem Grund wird bei der Isolierung von DNA aus (grampositiven) Bakterien die Lysozymbehandlung vor der Lyse durchgeführt.
David gegen Goliath: Plasmide erholen sich zuerst!
Bei Prokaryonten und Hefen sind Plasmidisolationen mindestens ebenso wichtig wie die Isolierung genomischer DNA. Obwohl sie aus den gleichen Bausteinen bestehen und beide doppelsträngig sind, können diese beiden DNA-Typen während des Isolationsverfahrens eindeutig getrennt werden. Niedermolekulare Plasmide lassen sich leicht aus hochmolekularer genomischer DNA reinigen, basierend auf ihrer Löslichkeit und Re-Annealing-Rate.
Die bevorzugte Methode zur Isolierung von Plasmid-DNA ist die alkalische Lyse, eine 1979 erfundene Technik (Birnboim & Doly, 1979). Sie funktioniert perfekt für konventionelle Plasmide mit einer Größe von weniger als 15 kbp. Das Bakterienzellpellet wird im Tris-Puffer mit EDTA und RNase A resuspendiert. Der Lysepuffer, der NaOH und SDS enthält, wird den homogenisierten Zellen zugegeben und die Lösungen werden durch sanfte Inversion gemischt. Für die Isolierung von Plasmid-DNA ist das Vorhandensein von chaotropen Salzen während der Lyse kontraproduktiv und verhindert die Trennung des Plasmids vom Chromosom. Im Gegensatz zum Lysepuffer für genomische DNA sollten der alkalischen Lyselösung also keine chaotropen Salze zugesetzt werden. Außerdem sollten umfangreiche Inkubationszeiten in der Lyselösung vermieden werden, da sie das Risiko einer irreversiblen Denaturierung der Plasmid-DNA bergen.
Die Inkubation von Zellen unter alkalischen Bedingungen führt zur Denaturierung sowohl von DNA-Spezies, Plasmiden als auch von genomischer DNA.
Die Inkubation von Zellen unter alkalischen Bedingungen führt zur Denaturierung sowohl von DNA-Spezies, Plasmiden als auch von genomischer DNA.
Während der Lyse wird die Lösung durch die Solubilisierung von Makromolekülen klarer und hochviskoser.
Durch Zugabe einer Kaliumacetat-/Essigsäurelösung wird das Gemisch neutralisiert und alle Makromoleküle mit Ausnahme von Plasmid-DNA ausgefällt. Dies wird erreicht, indem die kleine kreisförmige Plasmid-DNA schnell renaturiert. Im Gegensatz dazu bleiben die längeren, verwickelten genomischen DNA-Stränge getrennt, kleben mit denaturierten Proteinen zusammen und sind mit SDS-Molekülen beschichtet. Die Kaliumionen im Neutralisationspuffer ersetzen die Natriumionen des SDS und führen zur Bildung eines weißen, trüben Niederschlags (Ish-Horowicz & Burke 1981), der zusammen mit restlichen Zelltrümmern durch Zentrifugation sedimentiert wird. Die renaturierte Plasmid-DNA, die von der Vielzahl der intermolekularen Wechselwirkungen unbeeinflusst bleibt, bleibt im Überstand und wird weiter gereinigt.
Der entscheidende Punkt im Trennprozess ist es, eine starke Vermischung oder Wirbelbildung während der Lyse und Neutralisation zu vermeiden, da die genomische DNA leicht in kleine Fragmente zerfällt, die schließlich den plasmidhaltigen Überstand verunreinigen. Die DNA ist sehr resistent gegen chemische Einflüsse, aber je länger die Kette ist, desto empfindlicher ist das DNA-Molekül gegenüber physikalischen Kräften. Es ist unvermeidlich, dass die großen DNA-Chromosomen während des Reinigungsprozesses brechen, aber solange sie nicht in kleine Stücke zerlegt werden, ist dieser Bruch nicht von Nachteil. Auch das Gegenteil ist der Fall: Nachfolgende PCR-Reaktionen sind durch das bessere Schmelzen der gebrochenen DNA effizienter.
Die Endreinigung der Plasmidlösung kann mit den gleichen Methoden wie bei genomischer DNA durchgeführt werden, z.B. durch Phenol-Chloroformextraktion, Ethanolfällung oder mit einer Kieselsäule. Die letztgenannte Methode ist heute die beliebteste, weil sie sehr schnell ist und hochwertige Plasmid-DNA liefert, die sich für Transformation, enzymatische Verdauung, Mutagenese usw. eignet.
Methoden der DNA-Isolierung
Bei der Isolierung von DNA geht es natürlich darum, mit minimalem Zeit- und Kostenaufwand ein Maximum an Reinheit und Menge zu erreichen. Leider ist es selten möglich, all dies zu erreichen. Je nach Quelle, DNA-Typ (genomisch, Plasmid oder Organellen, z.B. Mitochondrien), Probengröße, Probenalter und erforderlicher Reinheit werden unterschiedliche Methoden der DNA-Isolierung eingesetzt. Aber nicht jede Methode ist für jeden nachgelagerten Prozess geeignet. Die wichtigsten DNA-Isolationstechniken sind kommerzielle Festphasen-Kits (Säulen mit Silica- oder Anionenaustauschmatrizen), Phenol/Chloroformextraktion und monophasische Reagenzien.
Die traditionelle Methode: Phenol-Chloroform-Extraktion
Die 1956 erstmals beschriebene Reinigung von Nukleinsäuren mit Phenol zur Entfernung hydrophober Verunreinigungen ist bis heute weit verbreitet.
Die Phenol-Chloroform-Extraktion ist billig, aber effektiv. Nach der Ethanolfällung zur Entfernung von Salz- und organischen Lösungsmittel-Verunreinigungen liefert das Verfahren hohe Mengen an reiner DNA. Andererseits ist das Verfahren zeitaufwändig, erfordert mehrere Transfers von Reaktionsröhrchen zu Reaktionsröhrchen, die das Risiko von Verunreinigungen (durch Fremdnukleinsäuren, aber auch durch Phenol/Chloroform-Verschleppung) tragen und beinhaltet gefährliche Chemikalien. Dennoch ist die Phenolextraktion oft die Methode der Wahl für sehr kleine und große DNA-Fragmente sowie für alte und teilweise abgebaute DNA.

Phenol als Reinigungsmittel für DNA
Basierend auf der Entdeckung, dass Phenol geeignet ist, Proteine aus wässrigen Lösungen zu extrahieren (Grassmann & Deffner, 1953), berichtete Kirby Phenol als Mittel zur Nukleinsäurereinigung (Kirby, 1956). Mit einer Phenol-Wasser-Mischung extrahierte er ein Gewebehomogenat und zeigte, dass RNA in der wässrigen Phase getrennt wurde, während Proteine, die mit DNA verbunden waren, in die Interphase transferiert wurden. Kurz darauf stellte Kirby fest, dass in Gegenwart bestimmter anionischer Salze (z.B. p-Aminosalicylat und Natriumbenzoat) beide Nukleinsäurearten, DNA und RNA, in der wässrigen Phase angereichert werden (Kirby, 1957). Bis auf wenige Anpassungen (anionische Detergenzien wie SDS ersetzten die anionischen Salze, Zugabe von Chloroform und Isoamylalkohol sowie Ausgleich des pH-Wertes) ist die Kirby-Methode heute noch zeitgemäß.
Polare, hydrophile Verbindungen wie DNA, RNA und Proteine lösen sich meist am besten in polaren Lösungsmitteln (mit Wasser als Lösungsmittel mit maximaler Polarität). Aber im Gegensatz zu Nukleinsäuren bieten Proteine auch eine Reihe von unpolaren Strukturen. Die unpolaren Seitenketten von Phenylalanin, Leucin, Isoleucin, Valin, Prolin, Methionin und Alanin ermöglichen es dem Protein, in Lösung zu bleiben, wenn es einem weniger polaren oder sogar unpolaren Lösungsmittel ausgesetzt ist. Die Proteine ordnen sich neu an und setzen die unpolaren Seitenketten der Oberfläche aus, während die geladenen und polaren Reste im Proteinkomplex verborgen sind. Diese Eigenschaften ermöglichen die Extraktion von Proteinen aus einer wässrigen Phase mit einem weniger oder sogar unpolaren Lösungsmittel. Phenol ist trotz seines elektronegativen Sauerstoffatoms deutlich weniger polar als Wasser, da der Phenylring die Elektronendichte über das gesamte Molekül verteilt, aber nicht auf das Sauerstoffatom konzentriert..
Für die DNA-Isolierung muss das Phenol mit Tris auf einen End-pH-Wert von >7,8 pH äquilibriert werden, um sicherzustellen, dass die DNA negativ geladen und damit in der organischen Phase unlöslich ist. Ausgehend vom Zelllysat wird ein gleiches Volumen an tris-gepuffertem Phenolchlorchloroform (1:1) oder tris-gepuffertem Phenolchloroform-isoamylalkohol (25:24:1) zugegeben und die Lösung durch Vortexen ("kleine DNA-Moleküle" von <10 kb), sanftes Schütteln (10-30 kb) oder langsames Umkehren oder Drehen (>30 kb) vermischt. Chloroform denaturiert effizient Proteine, vermeidet die Rückhaltung von Wasser in der organischen Phase und verbessert die Phasentrennung durch Erhöhung der Dichte der organischen Phase. Die Zugabe eines kleinen Volumens Isoamylalkohol reduziert die Schaumbildung während des Extraktionsprozesses und garantiert mit Blick auf die RNA-Isolierung die Deaktivierung von RNasen (Green & Sambrook, 2012).
Die Zentrifugierung beschleunigt die Trennung der beiden Phasen, so dass die wässrige Phase mit dem niedrigeren spezifischen Gewicht oben drauf entsteht. Aber Vorsicht: Ein hoher Salzgehalt oder ein hoher Anteil an Saccharose in der wässrigen Lösung kann zu einer Umkehrung der beiden Phasen führen! Es wird daher dringend empfohlen, darauf zu achten, dass die richtige Lösung weiterverarbeitet wird. Da äquilibriertes Phenol im allgemeinen 8-Hydroxychinolin als Stabilisator enthält, kann die organische Phase durch ihre gelbe Farbe identifiziert werden. Nach der organischen Extraktion befindet sich die DNA (und die RNA, wenn während der Lyse keine RNase A zugegeben wurde) noch in der wässrigen Phase, während denaturierte Proteine in die Zwischenphase eingetreten sind und Lipide in die organische Phase überführt wurden.
Nach wiederholten Extraktionen mit dem phenolhaltigen Lösungsmittel wird die DNA-haltige wässrige Phase mehrmals mit Chloroform (oder Chloroform/Isoamylalkohol) extrahiert, um das restliche Phenol zu entfernen. Schließlich wird die reine DNA aus einer Lösung mit Ethanol oder Isopropanol ausgefällt.
Produkte von Panreac AppliChem für die Phenol-Chloroform-Extraktion von DNA
| Prod. Nr. | Beschreibung | Kommentar |
| A1153 | Phenol equilibriert, stabilisiert 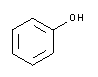 |
Reines Phenol ist eine farblose und kristalline Substanz. Verflüssigtes Phenol ist anfällig für Oxidation (besonders wenn der pH-Wert mit Tris ausgeglichen ist) und die phenolischen Oxidationsprodukte führen Strangbrüche in Nukleinsäuremoleküle ein und fördern die Vernetzung. Phenol-Lösungen sollten klar und farblos sein, rosa oder bräunliche Lösungen sollten entsorgt werden. Um Oxidation zu verhindern, wird dem verflüssigten Phenol häufig 8-Hydroxychinolin zugesetzt. Die Haltbarkeit von equilibriertem und 8-Hydroxychinolin-stabilisiertem Phenol beträgt ca. 9 Monate. Als positiver Nebeneffekt hemmt 8-Hydroxychinolin teilweise Ribonukleasen. |
| A1624 | Phenol wassergesättigt, stabilisiert | |
| A1578 | Phenol wassergesättigt, nicht stabilisiert | |
| A1594 | Phenol kristallin | |
| A0889 | Phenol equilibriert, stabilisiert : Chloroform : Isoamylalkohol 25 : 24 : 1 | |
| A3691 | Chloroform | |
| A2610 | Isoamylalkohol |
Die All-in-One-Lösung: Saure Guanidiniumthiocyanat-Phenol-Extraktion
Zugegebenermaßen zielt diese Technik nicht wirklich auf die DNA-Isolierung ab, aber dennoch ist es möglich, sie für diesen Zweck einzusetzen. Und da diese Technik interessante Möglichkeiten bietet, sollte sie in diesem Artikel erwähnt werden.
Die saure Guanidiniumthiocyanat-Phenol-Extraktion, die 1987 von Chomczynski & Sacchi zur Isolierung von RNA erfunden wurde, ermöglicht die Zelllyse und die aufeinanderfolgende Trennung von RNA, DNA und Proteinen mit einem Reagenz. Die Technik kombiniert die Wirkung von chaotropen Salzen auf die Struktur und Löslichkeit von Makromolekülen mit den Extraktionseigenschaften von saurem Phenol. Das unter den Namen Trizol®, TRI REAGENT® und TRItidy™ G vertriebene Reagenz von Chomczynski eroberte die Labors und gehört nach wie vor zu der ersten Wahl für die RNA-Isolierung.
Die Geburt monophasischer Reagenzien
Die Geschichte der von Chomczynski & Sacchi erfundenen sauren Guanidiniumthiocyanat-Phenolextraktion (oder der "einstufigen" Methode) ist in erster Linie die Geschichte der RNA-Isolierung. 1951 wurde erstmals die Verwendung von Guanidiniumchlorid in der RNA-Isolierung beschrieben (Volkin & Carter, 1951). Nach Kirby's Einführung von Phenol als Deproteinisierungsreagenz scheinen Guanidiniumsalze vorübergehend ihre Bedeutung zu verlieren, aber "Guanidiniumchlorid in der Isolierung von Nukleinsäuren" wurde in den 1960er Jahren wieder fokussiert (Cox 1968). 1979 veröffentlichten Chirgwin et al. ein Verfahren zur Isolierung nicht abgestufter RNA aus ribonukleasereichem Gewebe, bei dem Guanidiniumthiocyanat anstelle von Guanidiniumchlorid verwendet wurde. 1987 verwendeten Chomczynski & Sacchi zunächst eine Kombination aus Guanidiniumthiocyanat und Phenol-Chloroformextraktion zur RNA-Isolierung. Basierend auf dem gleichen Prinzip wurde die Methode für die gleichzeitige Isolierung von RNA, DNA und Proteinen erweitert (Chomczynski, 1993). Zwei Jahre später wurden weitere Modifikationen veröffentlicht, bei denen Bromchlorpropan (Chomczynski & Mackey, 1995a), das weniger toxisch ist als Chloroform und eine engere Interphase bildet, und Isopropanol (Chomczynski & Mackey, 1995b) zur verbesserten Isolierung von RNA aus Polysaccharid- und proteoglykanreichen Quellen verwendet wurden. Die Entwicklung konzentrierte sich schließlich auch auf die Isolierung genomischer DNA (Chomczynski et al., 1997).
Die einphasige Lösung enthält Wasser, Phenol, Guanidiniumthiocyanat (auch oft als Guanidinthiocyanat bezeichnet), β-Mercaptoethanol und ein Reinigungsmittel. Das chaotrope Salz Guanidiniumthiocyanat lysiert die Zellen, denaturiert die freigesetzten Makromoleküle und inaktiviert RNasen und andere Enzyme. Als Detergens eignet sich Laurinylsarcosin ("Sarkosyl"), da Laurin im Gegensatz zu SDS eine hohe Löslichkeit in chaotropen, salzreichen Puffern aufweist. Die Zugabe von Sarkosyl zu Zell- oder Gewebehomogenaten verbessert die Reinheit der durch Guanidiniumsalze isolierten RNA und reduziert das Schäumen während der Homogenisierung (MacDonald et al., 1987).

Nach der Lyse der Zelle im Reagenz (z.B. durch wiederholtes Pipettieren) wird Chloroform (oder alternativ Bromchlorpropan) zugegeben, was zur Bildung einer zweiten Phase führt. Während DNA und Proteine in der neu gebildeten organischen Phase und der Interphase angereichert sind, wird RNA selektiv in der wässrigen Phase zurückgehalten. RNA, DNA und Proteine werden durch Alkoholfällung isoliert.
Warum wird hydrophile DNA in die organische Phase überführt? Und wie ist es möglich, dass RNA dagegen nicht aus der Phenol-Chloroformmischung extrahiert wird? DNA und RNA scheinen sich auf den ersten Blick sehr ähnlich zu sein, und unter neutralen Bedingungen (pH 7-8) bleiben beide Moleküle wie erwartet in der wässrigen Phase. Die Verwendung von nicht äquilibrierten und damit sauren Phenollösungen ermöglicht jedoch die Anreicherung von DNA-Molekülen in der unpolaren organischen Phase. Warum verhalten sich diese beiden Arten von Nukleinsäuren unter sauren Bedingungen also unterschiedlich? Die Antwort liegt vor allem in den strukturellen Unterschieden zwischen DNA und RNA: Bei dem vorherrschenden pH-Wert von 4-5 werden die Phosphatgruppen der doppelsträngigen (und weniger sauren) DNA protoniert und die Affinität der nun ungeladenen (aber immer noch doppelsträngigen) DNA-Moleküle zum organischen Lösungsmittel steigt stark an. Das Phosphatrückgrat der einzelsträngigen RNA ist ebenfalls weitgehend protoniert, aber durch die Exposition der Purin- und Pyrimidinbasen ist die RNA in der Lage, Wasserstoffbrücken mit den umgebenden Wassermolekülen zu bilden (Zumbo, 2011). Dadurch verliert die RNA ihre hydrophilen Eigenschaften nicht und bevorzugt dennoch die wässrige Phase.
Monophasische Reagenzien von PanReac AppliChem: saure Guanidiniumthiocyanat-Phenolextraktion
| Prod. Nr. | Beschreibung | Kommentar |
| A4051 | TRItidy G™ | Gebrauchsfertige Lösung zur sequentiellen Isolierung von RNA, DNA und Proteinen. |
| A3418 | DNA - Isolierungsreagenz für genomische DNA | Phenolfreies, gebrauchsfertiges Reagenz zur Isolierung genomischer DNA aus menschlicher, tierischer (inkl. Mäuseschwanz), pflanzlicher, hefiger, bakterieller und viraler Herkunft.. |
Schnelle Entsalzung: Ethanolfällung
THeute wird die Ethanolfällung oft als letzter Schritt der DNA-Isolierung durch organische Extraktion oder Anionenaustauschchromatographie eingesetzt. Die Möglichkeit, DNA aus der Lösung auszufällen, ermöglicht nicht nur die Entfernung von alkohollöslichen Salzen, organischen Restlösungsmitteln und Reinigungsmitteln, sondern bietet auch die Möglichkeit, DNA zu konzentrieren.
In neutralen wässrigen Lösungen ist das negativ geladene Phosphat-Grundgerüst der DNA-Moleküle mit den hochpolaren Wassermolekülen gesättigt, die eine Interaktion mit kationischen Molekülen verhindern. Steigende Ethanolkonzentrationen führen zu einer Störung der Hydrathülle und ermöglichen die Bildung von ionischen Bindungen zwischen den Phosphatgruppen und positiv geladenen Ionen. In Lösungen, die mindestens 65 % Ethanol und eine ausreichende Menge an Kationen enthalten, werden daher die zuvor negativ geladenen DNA-Moleküle neutralisiert. Der Ladungsverlust minimiert die Abstoßungskräfte zwischen den Molekülen und lässt schließlich die DNA ausfallen. Die genaue Salzkonzentration ist entscheidend, um sicherzustellen, dass einerseits die gesamte DNA aus der Lösung gewonnen wird und andererseits die anionischen Salze nicht mit präzipitieren. Zur Verbesserung der Effizienz werden die Zielnukleinsäuren oft mit nukleasefreien, inerten "Trägern" wie Glykogen oder linearem Polyacrylamid Co-Prezipitiert.
Die häufigsten in Ethanolfällungen verwendeten Salze sind Ammoniumacetat, Natriumacetat und Natriumchlorid.
Eine Natriumacetatlösung bei einem sauren pH-Wert von 5,2 ist das Standardreagenz für die Nukleinsäurefällung. Natriumchlorid ist die erste Wahl, wenn in der Probe SDS enthalten ist, das seine Löslichkeit in Gegenwart von Alkohol sicherstellt und die Fällung von detergenzfreier DNA ermöglicht. Wenn die Probe mit dNTPs oder Oligosacchariden kontaminiert ist (einschließlich DNA-Proben, die aus Agarosegelen durch Agaraseverdau gewonnen werden), ist Ammoniumacetat am besten geeignet, da die Ammoniumkationen eine Co-Präzipitation dieser beiden Arten verhindern (Quelle: Green & Sambrook, 2012).
Isopropanol kann alternativ zur DNA-Präzipitation eingesetzt werden; es wird meist für große Mengen verwendet, da nur die Hälfte der Menge zur Entwässerung der DNA-Moleküle benötigt wird. Leider ist die Löslichkeit von Salzen in Lösungen mit 35% Isopropanol im Vergleich zu 65% Ethanol reduziert, was das Risiko von Salz-Co-Präzipitation erhöht. Darüber hinaus ist es weniger flüchtig als Ethanol und daher schwieriger zu entfernen, was das Risiko einer Verschleppung von Alkohol in die Endprobe erhöht.
Produkte von PanReac AppliChem zur Alkoholfällung von Nukleinsäuren
Die bequeme Methode: Handelsübliche Kits mit reinen Silica- und Anionenaustausch-Säulen
Spin-Kits auf Silica-Basis:
Die Fähigkeit der DNA, sich bei hohen Salzkonzentrationen unter alkalischen Bedingungen schnell und selektiv an Silikate zu binden, wurde erstmals 1979 beschrieben (Vogelstein & Gillespie, 1979): DNA wurde aus Agarosegelen entfernt und in Gegenwart von Natriumiodid an Glas gebunden.
Positiv geladene Ionen schützen die saure Siliziumdioxidoberfläche und fördern die Adsorption der DNA-Moleküle durch das negativ geladene DNA-Grundgerüst. Die Art des Brückenkations bestimmt die Bindungsstärke (Romanowski et al., 1991). Das Waschen mit chaotropen Salzlösungen entfernt restliche Proteinverunreinigungen, ohne die immobilisierte DNA zu beeinträchtigen. Nicht nur Salze, auch die Zugabe von Alkohol beeinflusst die Interaktion zwischen Matrix und DNA. Die kieselsäuregebundene DNA widersteht Waschverfahren mit 70%igen Ethanollösungen, die den Überschuss an Salzen, Metaboliten, RNA, Kohlenhydraten und anderen alkohollöslichen Biomolekülen verdrängen. Nur die Elution mit reinem Wasser oder salzarmen Lösungen (z.B. TE-Puffer) setzt die DNA frei. Denn der große Überschuss an Wassermolekülen ersetzt die ionischen Bindungen und rehydriert die DNA sowie die Oberfläche der Silica-Partikel.
Die Spin-Kits auf Silica-Basis sind sehr beliebt für die Isolierung genomischer DNA sowie für die Isolierung von Plasmiden. Die Reinheit der eluierten DNA ist hoch, und aufgrund der niedrigen Salz-Elutionsbedingungen ist eine weitere Entsalzung der DNA nicht erforderlich. Das Verfahren ist schnell, einfach durchzuführen und liefert reproduzierbare Mengen und Qualität. Leider sind die Säulen nicht für sehr kleine DNA-Fragmente geeignet. Je kleiner die DNA-Moleküle, desto enger ist die Bindung zwischen Silica-Matrix und DNA. Infolgedessen können kleine DNAs nicht effektiv aus der Säule gewonnen werden (Green & Sambrook, 2012). Ein weiterer Nachteil ist, dass die Bindungskapazität von Silica nur moderat ist. Daher sind diese "Mini"-Säulen nur für kleine Mengen an DNA geeignet, typischerweise bis zu 20 µg.
Die Bedeutung von chaotropen Salzen in Nukleinsäureisolierungsprozessen.
Chaotrope Substanzen wie Guanidiniumsalze, Harnstoff oder Lithiumperchlorat zeigen eine Reihe positiver Auswirkungen auf den Isolationsprozess:
1. Lyse der Zellmembran,
2. Denaturierung von Proteinen, DNA und anderen Makromolekülen,
3. Inaktivierung von Nukleasen,
4. Förderung der Nukleinsäurebindung an reines Silica-Material.
Alle diese Funktionen basieren auf der gleichen Wirkungsweise. Die chaotropen Substanzen stören intrazelluläre Interaktionen basierend auf Wasserstoffbrücken, hydrophoben Effekten und van der Waals-Kräften, was zur Denaturierung von Proteinen (wodurch die Proteinaktivität signifikant reduziert wird) und DNA, aber auch zur Reorganisation und schließlich zum Zusammenbruch der Membran führt. Die Ansammlung chaotroper Salze im hydrophoben Bereich der Lipiddoppelschicht gefährdet die Membranintegrität stark. Durch den Austausch der vorhandenen Wasserstoffbrücken mit benachbarten Wassermolekülen durch Salzbrücken zerstören die chaotropen Salze die Hydratationshülle, die die Nukleinsäuren umgibt, verringern deren Löslichkeit, maskieren Ladungen und vermitteln die Adsorption an Siliziumdioxidoberflächen.
Anionenaustausch-basierte Spin-Kits
Das Prinzip des Anionenaustausches unterscheidet sich völlig von der reinen DNA-Aufreinigung auf Silica-Basis. Im Gegensatz zur negativ geladenen Silikatoberfläche weist das Matrixmaterial eine hohe Dichte an positiven Ladungen auf. Das hydrophile Anionenaustauscherharz besteht aus großporigen Silica-Kugeln, die mit kationischen Gruppen beschichtet sind. Die maximale Bindung der DNA erfolgt unter leicht sauren salzarmen Bedingungen, die eine direkte und ungestörte ionische Interaktion der Phosphatgruppen der DNA mit der kationischen Oberfläche des Harzes ermöglichen.
Verunreinigungen werden durch Waschen mit mittleren Salzpuffern (~ 0,8 - 1,0 M NaCl, je nach pH-Wert) entfernt. Die DNA wird mit salzreichen Lösungen bei einem pH-Wert um 8 eluiert. Der Überschuss an Anionen ersetzt die gebundenen DNA-Moleküle und sättigt die Oberfläche der Anionenaustauschmatrix. Anionenaustauschmaterialien garantieren eine sehr reine DNA und bieten im Vergleich zu reinem Silica aufgrund ihrer hohen Ladungsdichte eine stark erhöhte Bindungskapazität. Daher werden Anionenaustauschmatrizen bevorzugt für großflächige DNA-Isolierungen eingesetzt. Nachteilig sind die Elutionsbedingungen, nämlich die Erfordernis sehr hoher Salzkonzentrationen, um die DNA aus der Säule freizusetzen. Die anschließende Entsalzung ist für nachfolgende Prozesse oft unerlässlich.
Säulen von kommerziellen Spin-Kits werden üblicherweise nach einmaligem Gebrauch entsorgt, da 5-10 % der DNA in der Matrix verbleiben. Natürlich ist das eine Verschwendung von Ressourcen und Geld. Und das ist nicht zwingend erforderlich, da die Matrix selbst noch perfekt funktioniert (Green & Sambrook, 2012). PanReac AppliChem bietet ein Produkt zur Regeneration von reinen Silica- und Anionenaustausch-Säulen an, welches die Bindungskapazität sogar noch verbessert. Die Säulenregenerationskits maxXbond™ basieren auf der ExitusPlus™-Technologie (siehe Kapitel Dekontamination), biologisch abbaubar und nicht gefährlich.
Säulenbasierte DNA- und RNA-Aufreinigung Spin-Kits von PanReac AppliChem [AX: Anion Exchanger; SI: Pure Silica].
| Prod. Nr. | Beschreibung | Kommentar |
| Aufreinigung von DNA und DNA-Fragmenten | ||
| A5193 | DNA Isolation Spin-Kit Agarose | Standard Gel Extraction Kit SI] zur Isolierung von DNA aus Agarosegelen |
| Dekontamination & Regeneration von Spin-Säulen | ||
| MB007 | maxXbond™ | Silica-Matrizen sind eine Schlüsseltechnologie für die Aufreinigung von DNA. Die schnelle Isolierung von reinen DNA-Proben ist heute für eine Vielzahl von molekularbiologischen Protokollen in Forschung und kommerziellen Anwendungen unerlässlich. Produkte mit Silica-Matrizen sind von hoher Qualität und hohem Wert. Ihr größter Nachteil ist, dass sie nur einmal verwendet werden können, da nach der Elution erhebliche Mengen an DNA an die Silica-Matrix gebunden bleiben und die Bindungskapazität reduziert wird. Um dieses Problem zu lösen, haben wir das erste Regenerationssystem für Silica-Matrizen entwickelt: maxXbond™ für reines Silica und maxXbond™AX für Anionenaustausch-Säulen. Zwei innovative Lösungen entfernen alle Nukleinsäuren und Fremdstoffe aus der Matrix und stellen die ursprüngliche Bindungsfähigkeit wieder her. |
Referenzen
- Arena, A. (2010) Invest. Sci. J. 2(2): 20-29
- Birnboim, H.C. & Doly, J. (1979) Nucleic Acids Res 7: 1513-1523
- Chirgwin, J.M., Przybyla, A.E., MacDonald, R.J. and Rutter, W.R. (1979) Biochemistry 18: 5294-5299
- Chomczynski, P. & Sacchi, N. (1987) Anal. Biochem. 162: 156-159
- Chomczynski, P. (1993) BioTechniques 15: 532-537
- Chomczynski, P. & Mackey, K. (1995a) Anal. Biochem. 225: 163-164
- Chomczynski, P. & Mackey, K. (1995b) BioTechniques 19: 942-945
- Chomczynski, P., Mackey, K., Drews, R. and Wilfinger, W. (1997) BioTechniques 22(3): 550-3.
- Cox, R.A. (1968) in Methods in Enzymology (Grossman L & Moldave K., Eds.), 12-B: 120-129, Academic Press, Orlando, Florida
- Grassman W. & Deffner, G. (1953) Hoppe Zeylers Z Physiol Chem 293: 89-98.
- Green, M.R. & Sambrook, J. (2012) Molecular Cloning: A Laboratory Manual, Fourth Edition, Cold Spring Harbour Laboratory Press, Cold Spring Harbour, New York.
- Ish-Horowicz, D. & Burke, J.F. (1981) Nucleic Acids Res 9: 2989-2998
- Kirby, K.S. (1956) Biochem J 64:405-408
- Kirby, K.S. (1957) Biochem J 66:495-504
- MacDonald, R.J., Swift, G.H., Przybyla, A.E. and Chirgwin J.M. (1987) Methods Enzymol. 152: 219-227
- Romanowski, G., Lorenz, M.G. and Wackernagel, W. (1991) Appl Environ Microbiol 57: 1057-1061
- Vogelstein, B. & Gillespie, D. (1979) Proc. Natl. Acad. Sci. USA 76, 615-619
- Volkin, E. & Carter, C.F. (1951) J. Am. Chem. Soc. 73(4): 1516-1519
- Zumbo, P. (2011) White paper. physiology.med.cornell.edu/faculty/mason/lab/zumbo/files/rna_phenol_chloroform.pdf