ITW Reagents proudly presents the DNA-ExitusPlusTM Technology
DNA-ExitusPlusTM & DNA-ExitusPlusTM IF
Reagent for the removal of DNA and RNA contaminationsProduct codes A7089 and A7409
Available literature:
InfoPoint
Product-Info
Brochure
Laboratory poster (ask your copy to your local distributor)
Description
DNA-ExitusPlus™ is a patented reagent for the removal of DNA and RNA contaminations from laboratory surfaces and equipment (1). The solution employs a mild and non-corrosive chemistry for a rapid non-enzymatic degradation of nucleic acids. Already short incubation times with DNA-ExitusPlus™ completely remove unwanted DNA and RNA from work surfaces and tools (1, 2).There are two different versions of DNA-ExitusPlus™ available: DNA-ExitusPlus™ (A7089) includes a color indicator to easily visualize the surface area covered with the reagent. DNA-ExitusPlus IF™ (A7409) is almost without color. Both solutions darken with time due to redox-active components contained in the solutions.
Note: There are no differences in the application protocols of DNA-ExitusPlus™ and DNA-ExitusPlus IF™! Therefore we do not name the IF form in the following instructions for use.
The unique characteristics of DNA-ExitusPlus™

- Catalytic and cooperative effects of the components cause a very rapid non-enzymatic, non-sequence-specific degradation of DNA and RNA molecules which makes all contamination go away.
- All components of DNA-ExitusPlus™ are readily bio-degradable and not harmful or toxic for humans.
- No aggressive mineralic acids or alkaline substances are used. Equipment and materials are not damaged or corroded even after prolonged incubation times.
- No toxic fumes.
- Elevated temperatures above approx. 50°C speed up the reaction and the activity.
Instructions for use
- The optimal incubation time for the decontamination of surfaces is 10 minutes and a temperature above 20°C. After incubation, residual DNA-ExitusPlus™ is wiped off with a paper towel. It is not necessary to additionally clean with sterile water thereafter. This is a new feature in comparison to the traditional decontamination solutions.
- After the solution is completely dried, there is no further decontamination reaction taking place. Hence, an incubation time longer than 30 minutes is not necessary and also not useful. In case of severe contaminations a second application of the solution is recommended for highest efficiency.
- For removal of unwanted, dried residual traces of the reagent, we recommend to remove these traces with sterile water or 10X TE buffer and a paper towel.
To decontaminate laboratory surfaces: Apply DNA-ExitusPlus™ directly to the lab surface. Incubate for 10 minutes. Wipe off residual DNA-ExitusPlus™ thoroughly with a paper towel (remove dried residues with sterile water / 10X TE buffer). It is not necessary to rinse with water.
To decontaminate laboratory apparatus: Generously apply DNA-ExitusPlus™ to a paper towel and wipe all exposed surfaces of the apparatus thoroughly. Dry with a clean paper towel. To clean small parts, briefly soak them in DNA-ExitusPlus™ and dry.
To decontaminate plastic and glass vessels: Add ample DNA-ExitusPlus™ to enable coating the entire surface of the vessel by swirling or vortexing. Discard the solution and dry. Rinse vessels thoroughly with distilled water and dry.
To decontaminate pipettors: Following manufacturer’s instructions; remove the shaft from the pipettor and remove seals and gaskets from the shaft. Soak the shaft for one minute in DNA-ExitusPlus™, rinse the shaft thoroughly with water, let dry and reassemble.
A new technology for nucleic acid decontamination
DNA amplification is one of the most commonly used techniques in the modern research laboratory. The presence of contaminating DNA in and around PCR workstations can lead to unwanted artifacts during amplification. Principally, there are two ways to make DNA not amplifiable:- by degradation of the DNA (e. g. by the addition of DNases or chemical destruction), or
- by modification of bases - leaving the DNA strand intact, but blocked for reading by polymerases.
Using a DNA strand break assay (designed by multiBIND biotec GmbH, Germany; see Fig. 1), it has been shown that not all DNA decontamination solutions on the market totally degrade DNA! DNA-ExitusPlus™ is an improvement over those products and causes both strand breakages as well as degradation. When used properly at the work area, it totally eliminates the amplification of non-target DNA (Fig. 3). DNA-ExitusPlus™ is a non-alkaline, non-corrosive and non-carcinogenic cleansing solution which is effective on all surfaces.
A severe disadvantage of conventional decontamination reagents is revealed in a new test for the corrosive potential of their components. For this purpose, different metal plates were incubated for 20 minutes with identical aliquots of the reagents. The selected metals are representative for equipment and materials found in laboratories. The result of this corrosion test is documented in figure 2, showing that all known commercial products contain aggressive chemical substances with corrosive, harmful or even toxic effects. These conventional reagents are known to contain azides, mineralic acids like phosphoric acid or hydrochloric acid, aggressive peroxides or strong alkaline substances like sodium hydroxide. Even after an incubation of only 20 minutes, irreversible damages of the metal surfaces are observed in many cases (see Figure 2). The newly developed solution DNA-ExitusPlus™ exhibits its unique characteristics especially in this corrosion test. For all metal surfaces tested, no damage or corrosion is observed after treatment. DNA-ExitusPlus™ was also tested under identical conditions on many different plastic surfaces without any indications of damage (data not shown).
The reagent is stable for at least 12 months and is heat stable.
(1) Esser, K.-H. et al. (2006) DNA decontamination: DNA-ExitusPlus™ in comparison with conventional reagents. Nature Methods 3, 151
(2) Arena, A. (2010) DNA-ExitusPlus™ versus standard bleach solution for the removal of DNA contaminants on work surfaces and tools. Investigative sciences journal 2, 20-29
Quality testing
Quantification of the DNA degradation by analytical agarose gel electrophoresis or PCRIt is getting more and more important to exactly control the DNA degradation after decontamination measures in laboratories.
If DNA is incubated with DNA-ExitusPlus™ or any other decontamination reagent and a sample of the reaction mixture is loaded onto an agarose gel without prior neutralization or denaturation, a quantitative determination of the DNA degradation won't be possible. Many fragments will stick together to form larger units, even after strand breakages (10 to 20 homologous basepairs will be sufficient to hybridize by forming larger units). This phenomenon of the hybridzation is observed for DNA fragments with sticky ends e.g. of the λ DNA size marker for gel electrophoresis. This is the reason why such a marker DNA is denatured before loading onto a gel.
The reagents of other suppliers frequently contain high concentrations of strong acids or bases. If you do not neutralize these reaction mixtures before loading onto the gel, you will observe a change in the color of the pH indicator bromophenol blue (transition interval at pH 2 - 4.6 from greenishyellow to blueviolet). If you load a non-neutralized sample onto a gel, you may observe the destruction of the slot by the chemicals. Ethidium bromide will be destroyed under these conditions and can therefore not be applied since it won't stain for DNA. The lanes on the gel would be clear, even if large quantities of unstained DNA are present. By neutralizing the sample with Tris buffer, the correct color of bromophenol blue will become visible. Depending on the composition of the decontamination reagent, neutralization has to be performed with either 100 mM Tris pH 12 or 100 mM Tris pH 3, respectively. In case of DNA-ExitusPlus™, the buffer capacity of the loading buffer is sufficient to buffer even an 1:1 mixture of the decontamination reagent and the sample.
When the neutralization of bromophenol blue shows the correct pH, the samples are denatured at 90°C for 2 minutes.
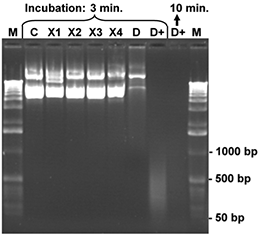
Aliquots of a CCC plasmid DNA (7 kb), 200 ng for each sample, were dissolved in 10 μl water and treated with 5 μl of one of the listed reagents for 3 or minutes, respectively, at room temperature. Finally, the samples were mixed with bromophenol blue loading buffer and denatured for 3 minutes at 92°C. The denatured samples were placed on ice and the complete reaction mixture was loaded onto an 1 % agarose gel. After gel electrophoresis the agarose gel was stained with ethidium bromide and photographed. The control (C) contains the intact CCC plasmid DNA (200 ng) after treatment with 5 µl of sterile water. Nicks and damages of the DNA strands generate fragments of lower molecular weight. These smaller DNA fragments can be identified in the gel by comparison with the control sample and the molecular weight marker (M; 1 kb ladder). The products X1-X4 cause very little degradation of the test DNA only, while product D (conventional DNA-Exitus™ ) was more effective under these conditions. Only DNA-ExitusPlus™ (D+) causes very rapid and nearly complete DNA degradation after 3 minutes. Thereby only residual DNA fragments smaller than 500 base pairs are observed. Prolonged incubation (10 min.) destroyed all plasmid DNA.
The high concentration of chemicals in the reagents of other suppliers leads to severe problems during PCR analysis. Samples collected by wiping of benches treated with such reagents did contain chemicals in such high concentrations that PCR was inhibited - even after dilution. Therefore, a positive control of the PCR with a mixture of a defined template and an aliquot of the sample collected is required to exclude false negative results. In our hands such a negative control-PCR still contained ingredients of the reagents at inhibitory concentrations making a further dilution or neutralization necessary.
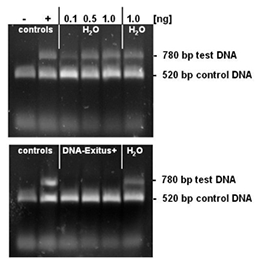
Selected amounts (0.1 to 1 ng) of a test DNA were lyophilised on the inner surface of PCR tubes. The PCR tubes with the lyophilised DNA sample were incubated for 20 seconds with either sterile water or DNA-ExitusPlus™ . Thereafter the tubes were washed twice with 100 µl of sterile water. For the PCR test reaction, mixtures of 50 µl were added to each tube. This reaction mixture contains primers for the amplification of the control DNA (control) and the test DNA. The control DNA (1 ng) is added into each sample and proves that the PCR reaction is not inhibited. Amplification of a DNA band corresponding to the test DNA indicates that intact DNA molecules of this template are still present. Upon complete degradation and removal of the test DNA the PCR reaction should not amplify any DNA fragment for this template. After electrophoresis through a 1% agarose gel the gel was stained with ethidium bromide and documented. The negative control with sterile water (H2O) exhibits DNA bands for the test and control templates. The PCR reaction after treatment with DNA-ExitusPlus™ amplifies only the fragment of the control DNA. This demonstrates that the treatment with DNA-ExitusPlus™ destroys and removes all traces of the test DNA template.

For this test metal plates were chosen that are typical for laboratory materials and equipment. Aliquots of 10 μl from each reagent listed were applied on the different metal surfaces. Sterile water was used as a control (0). After an incubation time of 20 minutes the reagents were wiped off and the metals were briefly washed with sterile water. After complete drying the metal plates were photographed. The reagents X2, X3 and D (see figure 2) for DNA decontamination cause irreversible corrosion and damages to many of the metal surfaces. For DNA-ExitusPlus™ (D+) no damage of any of the surfaces is observed. In some cases one observes a polishing effect by the removal of dirt or oxide layers.
Degradation of small DNA fragments
The strand breaking activity of DNA-ExitusPlus™ is independent of the size of the DNA fragments, since the destruction is based on a chemical action and not an enzymatic activity. Therefore, a 750 bp PCR product has been incubated for varying times with DNA-ExitusPlus™ (see Fig. 4). As expected, the primers cannot be detected even after the shortest incubation time. After 5 minutes of incubation with DNA-ExitusPlus™ almost all DNA is destroyed. We want to make clear why it is so: Assuming that a theoretical nicking activity of 100,000 nicks per minute is present, all DNA fragments will be destroyed, independent of their size. Smaller fragments will disappear first, than the larger ones. Therefore, transferring this theory to a test molecule (ccc form, 6 kb plasmid), after 5 minutes there will remain a small fraction of fragments with 200 to 500 bp in size only. The nicks will be introduced statistically at any site, leaving not a single class of fragments. Therefore, PCR analysis will be negative. When spraying DNA-ExitusPlus™ on surfaces in the lab, an enormous excess of approximately 1 to 5 ml reagent will be applied to the smallest quantities of DNA.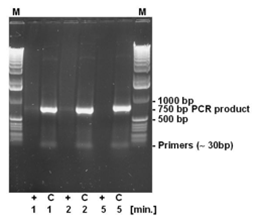
For testing the degradation of small DNA fragments, 500 ng DNA of a PCR reaction yielding a 750 bp PCR fragment has been incubated with DNA-ExitusPlus™ for the periods indicated (1, 2 and 5 minutes). After teatment, the DNA was denatured at 95°C for 2 minutes. (+) 5 µl DNA plus 5 µl DNA-ExitusPlus™ ; (C) control 5 µl DNA plus 5 µl water; (M) molecular weight marker 1 kb ladder.
Degradation of RNA by DNA-ExitusPlus™
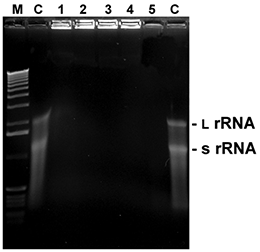
Fig. 5. RNA degradation
5 µl with 1 µl of total RNA from E. coli were mixed with 5 µl of the listed reagents and incubated at RT for the time given. Thereafter, samples were mixed with loading buffer, heated for 2 min. at 60°C and immediately loaded onto a 1.5% agarose gel with formamide/ formaldehyde as denaturing agents. After washing and renaturation of the gel RNA was visualized by EtBr staining (L rRNA: large ribosomal RNA; s rRNA: small ribosomal RNA).
M: 1 kb ladder
C: control (sterile water)
1:D+ 0.5 minute
2:D+ 1 minute
3:D+ 2 minutes
4:D+ 5 minutes
5: RNaseA (10 ng / 5 minutes)
Enhanced Efficiency by Increased Incubation Temperature
It is extremely difficult to remove and destroy DNA, dried to any surface or DNA "protected" by e.g. the envelope of a virus. Even autoclaving fails in some cases to degrade such DNA. The complete genome of an avian virus could be detected after autoclaving [Elhafi, G., Naylor, C.J., Savage, C.E. and Jones, R.C. (2004). Microwave or autoclave treatments destroy the infectivity of infectious bronchitis virus and avian pneumovirus but allow detection by reverse transcriptase-polymerase chain reaction. Avian Pathology 33, 303-306]. The activity of DNA-ExitusPlus™ as an additive to solutions during autoclaving has been tested. It could be shown, that increased temperatures improved the DNA-degrading activity of DNA-ExitusPlus™ !
50 ml cultures of recombinant E. coli strains where autoclaved at 120°C and 1.2 bar for 20 minutes with the addition of equal volumes of either water (-) or DNA-ExitusPlus™ (+). Subsequently, 10 µl aliquots of these cultures were investigated by analytical DNA agarose gels. The sample with sterile water added (-) shows large quantities of high molecular weight DNA fragments. Addition of the same volume of DNA-ExitusPlus™ (+) leads to the degradation of the DNA, showing fragment sizes below 20 base pairs.

The autoclaved recombinant E. coli cultures from fig. 5 contain a plasmid bearing the ampicillin resistance gene (AmpR-Gen). Aliquots (2 µl ) of the cultures were tested in a PCR reaction with primers for the complete AmpR gene after autoclaving. The sample with sterile water (-) results in a strong band of the complete AmpR gene (930 bp), while samples treated with DNA-ExitusPlus™ (+) prevented a positive result. As a positive control (K), a 2 µl aliquot of the sample with DNA-ExitusPlus™ was mixed with 2 ng template DNA coding for the AmpR-Gen. The amplification of the corresponding band shows that the PCR reaction does work under the assay conditions.
FAQs
What is the suggested contact time?For contaminations with small amounts of DNA 5 to 10 minutes at room temperature are suffcient. For higher amounts of DNA it is recommended to repeat the procedure after wiping off DNA-ExitusPlus™ .
In addition, the increase of temperature of the solution to 50°C or 60°C increases the degradation rate of DNA substantially.
Do any methods exist for detecting residuals of the component materials of DNA-ExitusPlus™?
The new formulation of DNA-ExitusPlus™ contains a low amount of color indicator that can be detected on surfaces after drying. In case any small residual traces of DNA-ExitusPlus™ have to be removed, they can easily be wiped off the surfaces with filter paper soaked in sterile water.
For inactivation of larger residual amounts of DNA-ExitusPlus™ the surfaces can be treated with a solution of sterile 10X TE buffer, pH 8.0. Make sure to prevent new contaminations during this treatment.
Your equipment includes polypropylene, stainless steel, EPDM and silicone gaskets. What information do we have in terms of "coating" or sticking to these materials?
EPDM = Ethylene-Propylene-Diene-Monomer
Polypropylene, stainless steel, EPDM and silicone gaskets are not damaged by DNA-ExitusPlus™ and the solution can easily be washed away by sterile water or 10X TE buffer, pH 8.0. Residual amounts of DNA-ExitusPlus™ have dried on these surfaces and can easily be removed by a single washing step with the same solutions listed and wiping off the solution with clean paper towels.
Thin tubes - how can I remove residual DNA-ExitusPlus™ ?
DNA-ExitusPlus™ has the advantage over other commercial products especially for tubes with a small diameter, because the ionic strength of all components are substantially lower than for other commercial products.
In addition all components of DNA-ExitusPlus™ are highly soluble in water and do not change the viscosity of water substantially. They all have a very low affinity for metal or plastic surfaces.
Therefore, even for tubes with a very small diameter one rinsing step with 10X TE buffer, pH 8.0, and a final rinsing with sterile water is enough for the complete removal of DNA-ExitusPlus™ .
As a final control, determine the pH of the sterile water after the final rinsing. A pH between 6 and 8 is acceptable. Please check the pH of the sterile water before the application. Caution: sterile water prepared with anion exchange resins can exhibit low pH.
Why do you see a bright fluorescence in the slots of DNA-ExitusPlus™ treated samples from fig. 5?
The fluorescence observed in the slots of the agarose gel is caused by ingredients of residual DNA-ExitusPlus™, but not DNA or RNA. These small molecules diffuse in all directions into the gel matrix despite electrophoresis. By contrast, DNA would accumulate at the bottom of the slot, in case it would precipitate. Under the experimental conditions chosen, neither RNA nor DNA would precipitate and the RNA samples of this experiment don’t contain high molecular weight DNA retained in the slots.