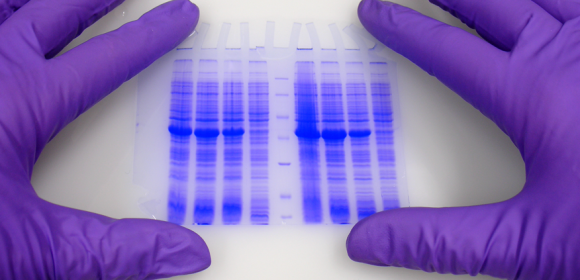

En Ciencias de la Vida el estudio de las proteínas es uno de los campos más importantes.
La expresión de lo que está codificado en nuestros genes se puede ver, y hoy en día también se recoge, bajo el término de Proteómica. Se han desarrollado muchas técnicas diferentes en los últimos años y décadas. Una de las más conocidas y utilizadas es la técnica PAGE de Electroforesis en Gel de Poliacrilamida.
Electroforesis en Gel de Poliacrilamida, PAGE
La electroforesis en gel de poliacrilamida se utiliza normalmente para la separación de proteínas, pero también el ADN (especialmente para visualizar las diferencias entre pequeños fragmentos) puede separarse mediante PAGE. La matriz está formada por filamentos de acrilamida reticulados con N,N-metilenobisacrilamida. ITW Reagents ofrece reactivos y mezclas preparadas para SDS-PAGE, PAGE nativo, DNA-PAGE desnaturalizante y no desnaturalizante, así como para geles de secuenciación.
Mezclas de Acrilamida
La capacidad de separación de un gel de poliacrilamida se determina por la proporción de mezcla de acrilamida y bisacrilamida. Cuanto menor sea la proporción de estos dos componentes en la mezcla, mayor será el grado de reticulación. Esto significa que un gel al 6% preparado a partir de una solución madre con una proporción de mezcla de 29 : 1, tiene un mayor grado de reticulación que un gel al 6% preparado a partir de una solución madre con una proporción de mezcla de 37,5 : 1.
Para la mayoría de las aplicaciones se utiliza una proporción de acrilamida: bisacrilamida de 29 : 1 o 37,5 : 1 (para la separación electroforética de ácidos nucleicos o proteínas). La proporción de mezcla de 19:1 es la solución elegida para la secuenciación de ADN. La preparación se simplifica utilizando soluciones madre de acrilamida acuosa al 30% o 40% con la proporción deseada.
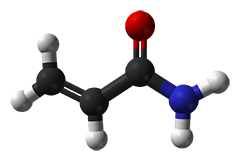
Las soluciones de acrilamida se denominan a menudo "estabilizadas con gas" y ofrecen una mayor vida útil. El "gas" es simplemente oxígeno, que tiene por objeto evitar una polimerización espontánea de las soluciones de acrilamida. Sin embargo, como los iniciadores de la polimerización APS y TEMED se añaden en exceso, la "desgasificación" (realizada en parte en el laboratorio antes de la iniciación de la polimerización) es en realidad innecesaria.
La polimerización espontánea no deseada es iniciada por radicales que típicamente se originan a partir del ácido acrílico. Las calidades más pobres a menudo contienen trazas detectables de ácido acrílico y las soluciones basadas en estos grados conllevan un alto riesgo de polimerización espontánea. En contraste, los grados recristalizados cuatro veces ("4K"), sin embargo, están libres de ácido acrílico. Para nuestro grado de biología molecular sólo se emplea acrilamida 4K especialmente probada para la ausencia de DNasas, RNasas y proteasas.
A bajas temperaturas de 2-8°C, el intercambio de oxígeno se reduce dentro de la solución de acrilamida y se facilita la polimerización espontánea. Por lo tanto, se recomienda su almacenamiento a temperatura ambiente. La siguiente tabla enumera las mezclas de acrilamida más comunes; hay más soluciones de acrilamida disponibles o bajo pedido.
Uso de mezclas de acrilamida
Las soluciones madre de acrilamida se diluyen para obtener la concentración de monómero deseada. El volumen total de la cantidad necesaria de solución de gel (por ejemplo, 100 ml) se divide por el contenido de la solución (por ejemplo, 30%) y se multiplica por la concentración final deseada (por ejemplo, 6% de acrilamida) para obtener el volumen necesario de la solución madre:
(100 ml / 30%) * 6% = 20 ml
Para preparar 100 ml de un gel de acrilamida al 6%, se añaden a la mezcla de gel 20 ml de la solución madre de acrilamida al 30%.
Por supuesto, también puede solicitar nuestras mezclas de acrilamida en polvo. Pero cuidado, el monómero de acrilamida es una fuerte neurotoxina que se acumula! Por lo tanto, se deben usar guantes y una máscara facial cuando se manipule acrilamida cristalina.

Soluciones tampón, SDS, APS, TEMED
El tampón de electroforesis más utilizado para SDS-PAGE es la SDS-trisglicina, el llamado tampón de Laemmli. Una alternativa es el sistema tris-tricina-SDS inventado por Schaegger & Jagow. Para los geles proteicos nativos, el tampón de tris-glicina es la primera opción.
Para iniciar la polimerización se añaden 100 µl de APS al 10% y 5-10 µl de TEMED por cada 10 ml de solución de gel. Dado que la polimerización es muy rápida inducida por TEMED y el iniciador radical APS, el gel debe ser vertido inmediatamente. El enfriamiento de la solución desacelera el proceso de polimerización. El APS (persulfato de amonio, A1142) no es muy estable en solución acuosa (normalmente se prepara una solución madre al 10% en agua), pero por experiencia, la solución puede almacenarse a 2-8°C durante varias semanas, o a -20°C durante meses sin perder su actividad.
TEMED (tetrametiletilendiamina, A1148) mejora la polimerización de acrilamida y bisacrilamida al catalizar la formación de radicales libres de APS. Se utiliza en una concentración de 50 µl / 100 ml de solución de gel. La estabilidad de TEMED es muy alta (2 años), si se evita el contacto con el agua.
Componentes tampón y otros productos químicos para la electroforesis en gel de poliacrilamida de proteínas
| Cód. Prod. | Descripción | Observaciones |
|---|---|---|
| A1142 | Amonio Peroxodisulfato (APS) BioChemica | APS; la concentración final recomendada en geles de acrilamida es de 0,1% (p/v). Preparar una solución madre al 10% (p/v) para una fácil aplicación. |
| A1148 | TEMED | N,N,N',N'-Tetrametiletilendiamina; la concentración final recomendada en geles de acrilamida es de 0,1% (v/v). |
| A0676 | SDS-Solución 10% para biología molecular | Sodio Dodecilo Sulfato; también disponible como solución al 20% (A0675) o cristalino (p. ej. A2572). |
Soluciones de Acrilamida listas para usar para SDS-PAGE
Ahorre tiempo y asegure resultados reproducibles: Nuestras soluciones de gel listas para usar para SDS-PAGE son su producto preferido para una preparación de gel fácil y rápida. ITW Reagents ofrece soluciones listas para usar para apilamiento y resolución de gel según Laemmli, basadas en una relación acrilamida/bisacrilamida de 29 : 1 con 0,1% de SDS y Tris.
Para preparar el gel de acrilamida, 100 ml de cada solución lista para usar (véase el cuadro siguiente) se completan con 1 ml de APS (de una solución madre al 10% a base de A1142)) y 50 µl de TEMED (A1148) para iniciar la polimerización del gel. Para la electroforesis se utiliza un tampón de SDS-trisglicina (por ejemplo, A1415).
Marcadores de proteínas y colorantes de proteínas
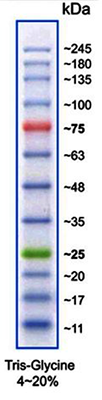
(Imagen: Marcador de proteína VI)
Patrones de tamaño molecular de proteínas para SDS-PAGE
| Cód. Prod. | Descripción | Observaciones |
| A8889 | Marcador de Proteínas VI (10-245) preteñido | 12 bandas; marcador listo para usar y preteñido para una mejor detección y orientación |
Para la tinción no específica de las bandas de proteínas en el gel de poliacrilamida se utiliza principalmente Coomassie® Brilliant Blue R-250. Si se requiere una sensibilidad más alta, se utiliza tinción de plata nitrato. Antes o durante el procedimiento de tinción, las proteínas deben fijarse en la matriz de gel. Esto se hace mediante una mezcla acuosa de etanol y ácido acético o ácido tricloroacético que conduce a la desnaturalización de las proteínas y a la precipitación. Para la fijación de pequeñas proteínas básicas, el formaldehído es más adecuado.
Colorantes proteínicos para la tinción de geles de poliacrilamida
| Cód. Prod. | Descripción | Observaciones |
| A1092 | Coomassie® Azul Brillante R-250 | Uno de los colorantes más utilizados para proteínas en SDS-PAGE. El complejo proteína-tinte tiene un máximo de absorción a 549 nm. Por cada aminoácido cargado positivamente se unen aproximadamente de 1.5 a 3 moléculas de Coomassie® Azul Brillante R-250. La sensibilidad es de alrededor de 200-400 ng de proteína/banda. |
| A3480 | Coomassie® Azul Brillante Blue G-250 | La principal aplicación de este colorante es el ensayo de Bradford (determinación de la concentración de proteínas), pero también es posible la tinción de proteínas en geles de poliacrilamida. |
| A3930 | Rodamina B 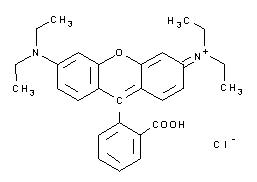 |
Ver Negro de Eriocromo T para análisis, ACS (131439). |

La historia de la electroforesis - pasado y presente
Las moléculas cargadas migran en un campo eléctrico. Este es el principio básico de la electroforesis, un método utilizado hoy en día en casi todos los laboratorios de biología y fundamental para la mayoría de las técnicas de separación y métodos analíticos.Cuando oímos el término electroforesis probablemente pensaremos en electroforesis en gel, agarosa o acrilamida, especialmente SDS-PAGE - que es hoy en día el método electroforético más popular y más ampliamente utilizado en la investigación en todo el mundo. Pero el campo de los métodos y aplicaciones electroforéticas es muy amplio y, a diferencia de las técnicas que dominan actualmente, los primeros pasos de la electroforesis se realizaron en solución libre sin ningún tipo de medio de apoyo.
Los primeros años
¿Cómo comenzó esta exitosa historia? La teoría básica de la electroforesis se desarrolló hace más de 200 años, pero fue sólo en la década de 1930 que Tiselius presentó la llamada electroforesis de límite móvil. Esta técnica de electroforesis de solución libre era adecuada para estudiar la movilidad de las moléculas cargadas. En el "aparato de Tiselius" los "límites móviles" formados por proteínas electroforéticamente migratorias fueron medidos por los cambios en la absorción de la luz o el índice de refracción. A diferencia de los métodos actuales, nunca se logró una separación completa de los componentes de una mezcla, independientemente de la duración del experimento. Sólo fue posible un análisis parcial de los compuestos de migración más rápidos y más lentos.
En la década de 1950, la electroforesis de límites móviles fue superada por la electroforesis de zona, un principio diferente de electroforesis que se describió por primera vez en 1939. En la electroforesis de zona, las proteínas, nucleósidos, aminoácidos y otras moléculas se separaron físicamente entre sí con la ayuda de un medio de soporte. Al principio se utilizaron papeles de filtro, pero pronto el método se mejoró considerablemente con soportes alternativos como el acetato de celulosa (Kohn 1957), el almidón (Smithies 1955), la poliacrilamida (Raymond & Weintraub 1959) y la agarosa (Hjertén 1961). El medio de soporte evita la sedimentación de las moléculas y, a diferencia de la electroforesis de límite móvil, permite una separación completa.
La invención de PAGE
Con la década de 1960, comenzó una nueva era en el campo de la electroforesis. Se introdujeron nuevas técnicas y se mejoraron en gran medida los métodos ya existentes. Se desarrolló la electroforesis de disco y el isoelectroenfoque. El Coomassie® Azul Brillante se utilizó inicialmente como colorante para visualizar bandas de proteínas después de la electroforesis en gel y se identificó la SDS para "enmascarar" la carga neta de proteínas durante la electroforesis de poliacrilamida.
Cinco años después de que Raymond & Weintraub introdujera los geles de poliacrilamida para la electroforesis de proteínas, este material fue utilizado por Ornstein y Davis como soporte para una técnica que es conocida y aplicada en casi todos los laboratorios bioquímicos hasta ahora: En 1964, desarrollaron la electroforesis de disco (discontinua), que combina la electroforesis de zona con la isotacoforesis.
La isotacoforesis se realiza en un sistema tampón discontinuo
Por isotacoforesis - inicialmente denominada método de migración iónica (Kendall & Crittenden 1923) o electroforesis por desplazamiento (Martin 1942) - se pueden generar límites precisos entre los componentes de la muestra. A diferencia de la electroforesis de zona, la isotaforesis se realiza en un sistema de amortiguación discontinua, compuesto por un electrolito principal de alta movilidad y un ión de terminación o de seguimiento de baja movilidad. Después de configurar el campo eléctrico, las moléculas migran a diferentes velocidades y, por lo tanto, se separan completamente unas de otras formando pilas; la molécula con la mayor movilidad sigue directamente al ión principal, mientras que las moléculas con la menor movilidad migran directamente delante del electrolito de terminación. Nada especial hasta ahora. Sin embargo, los iones de movimiento más rápido bajarán el campo eléctrico circundante mientras que los más lentos crearán un campo más alto. Como consecuencia, después de la separación, todas las moléculas migran con la misma velocidad (y esto es lo que significa literalmente el nombre de isotaforesis: migración a igual velocidad): los iones que migran más rápidamente serán retardados por el campo más débil circundante; los iones que migran más lentamente serán acelerados por el campo más fuerte a su alrededor. Por esta razón, el sistema tiene un efecto de autoafilado; tan pronto como un ión se difunde fuera de su propia banda, se desacelera o se acelera, forzándolo a retroceder a su banda original.
Volvamos al principio de la electroforesis de disco. Mediante el uso de 2 áreas diferentes de separación (apilamiento y resolución de gel) y un sistema de amortiguación discontinua, Ornstein y Davis previnieron la formación de agregados de proteínas durante la entrada en la matriz de gel y apoyaron la separación en bandas bien definidas.
El gel de apilamiento se caracteriza por un pH más bajo y poros más grandes que el gel de resolución adyacente.
El gel de apilamiento se caracteriza por un pH más bajo y poros más grandes que el gel de resolución adyacente. Ambos geles contienen sólo iones de cloruro (con Tris como contra-ión), mientras que el tampón del electrodo contiene sólo glicina. Al principio, las proteínas son "apiladas" y concentradas por el principio de isotacoforesis. Debido a los grandes poros del gel de apilamiento, el tamaño de la molécula no influye en su movilidad. La carga neta de glicina es casi nula en el pH del gel de apilamiento, por lo que la glicina funciona como ión terminal. Cuando el frente de la proteína llega al borde del gel de resolución de malla cerrada, las pequeñas moléculas de glicina pasan a través de las proteínas, entran en el área de resolución, se cargan más y se mueven junto con los iones de cloruro delante de la fracción de la proteína. Tan pronto como las proteínas están rodeadas por el tampón homogéneo comienzan a separarse según los principios de la electroforesis de zona: ahora su movilidad depende de su carga y tamaño, lo que finalmente conduce a un reordenamiento de la clasificación de proteínas.
Cuando Laemmli publicó en 1970 su famoso artículo sobre la separación de proteínas de fagos T4, utilizó el sistema tampón de Tris-Glicina-Cloruro de Ornstein y Davis. Pero en lugar de realizar una PAGE "nativa", añadió la SDS, que fue introducida por Shapiro y otros en 1967. En SDS-PAGE, el proceso de separación ya no se basa en la carga neta específica de las proteínas, sino en sus diferencias de peso molecular. La combinación de SDS-PAGE con el tampón "Laemmli" es la técnica más utilizada para la separación de proteínas hasta la fecha.
Otro hito en el desarrollo de la electroforesis en gel fue la separación isoeléctrica de proteínas. La teoría del enfoque isoeléctrico fue introducida por Svensson ya en 1961, pero sólo después de que Vesterberg sintetizara con éxito los anfolitos portadores para la generación de un gradiente continuo de pH, nació la técnica del enfoque isoeléctrico. En 1975, O'Farrell combinó el enfoque isoeléctrico con SDS-PAGE; la llamada electroforesis 2D (o "mapeo de proteínas", ya que cada proteína se caracteriza por una posición específica debido a su carga intrínseca y masa) se convirtió en una herramienta importante para el análisis de proteínas.
Un salto cuántico en la detección de proteínas en geles de acrilamida (especialmente en la electroforesis 2D) fue la introducción de las técnicas de tinción de plata por Merrill y otros en 1979. En comparación con la tinción predominante de Coomassie®, la sensibilidad se incrementó considerablemente, pasando de microgramos a nanogramos. En el mismo año, Towbin y otros realizaron el primer Western Blot; transfirieron proteínas separadas por SDS-PAGE a una membrana de nitrocelulosa.
SDS-PAGE: ¿Hay algo después de Laemmli?
El rendimiento de SDS-PAGE, acuñado con Laemmli, está presente hasta ahora, así que tenemos que preguntarnos si no hay alternativas. ¿Es éste el fin de la evolución de SDS-PAGE? ¿No ha habido más técnicas o mejoras en los últimos 40 años? - ¡Claro que hay algunos!
Existen algunas clases de proteínas que se comportan de manera anómala en SDS-PAGE: glicoproteínas, proteínas fuertemente básicas (de carga positiva) y algunas proteínas transmembranas hidrofóbicas. En cuanto a las glicoproteínas altamente hidrofílicas, el uso del tampón alcalino Tris-borato-EDTA (Poduslo 1981) proporciona una solución. Para separar las histonas, Panyim & Chalkley desarrollaron geles de poliacrilamida de urea ácida (geles AU) en 1969. Una alternativa es el gel TAU, que además contiene el detergente no iónico Triton®. Los geles TAU son adecuados para la identificación de modificaciones de proteínas como la acetilación y la fosforilación. Otras proteínas altamente cargadas pueden separarse según su tamaño utilizando el detergente catiónico bromuro de cetyltrimethylammonium en lugar de SDS (Eley y otros en 1979).
Para una mejor separación de los péptidos pequeños, Schägger & Jagow utilizó en 1987 un sistema tampón Tris-Tricina alternativo para SDS-PAGE. Cuatro años más tarde, los mismos investigadores introdujeron un sistema electroforético discontinuo para el aislamiento de proteínas de membrana de geles de acrilamida. En su electroforesis nativa azul, Coomassie en lugar de SDS se utiliza para inducir un cambio de carga en las proteínas. Además, el ácido aminocaproico y los detergentes no iónicos como Triton® X-100 sirven para mejorar la solubilización de las proteínas de la membrana. Los complejos de proteína-detergente resultantes se separan dentro de un PAG que contiene ácido aminocaproico y, para estudios sobre la estructura cuaternaria, se resuelven en los polipéptidos individuales mediante Tricine-SDS-PAGE de segunda dimensión. Para las proteínas de membrana pequeñas, se propone sustituir Coomassie® por taurodeoxicolato.
La belleza de la simplificación - Ahn Geles Simples
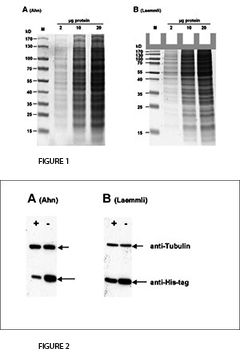
En 2001, Ahn et al. introdujeron una variante simplificada del procedimiento SDS-PAGE que consiste en un solo "gel único" con una mayor longitud para el gel de separación. En los geles de Tris-Glicina de Laemmli, la glicina sirve como el ion de movimiento lento. En contraste, los geles simples Ahn emplean tres aminoácidos para este propósito, a saber, glicina, serina y asparagina. En el protocolo original de Laemmli, el gel separador funciona con un valor de pH de 8,8, mientras que el gel único de Ahn funciona con un pH de 7,4. Con un valor de pH alcalino tan suave, la hidrólisis de acrilamida se minimiza. Mejora la estabilidad y la vida útil de las soluciones de acrilamida y geles. Los geles Ahn no contienen ningún detergente, pero se añade SDS al tampón de funcionamiento para permitir condiciones de desnaturalización durante la electroforesis. En el artículo original de Ahn y otros no se muestra ninguna comparación con otras variantes de SDS-PAGE. Queríamos estudiar las diferencias en el rendimiento de los geles según Ahn o Laemmli. Para ello se realizó electroforesis en gel sobre geles de poliacrilamida al 10 % según ambos protocolos. Además, investigamos la transferencia de proteínas a las membranas secantes en un ensayo de Western blot. Nuestros resultados en resumen son:
1. Electroforesis
La primera diferencia clara entre los geles Laemmli y Ahn es la longitud del gel de separación para el mismo tipo de casete de gel. Los geles simples según Ahn hacen geles de separación más largos (Fig. 1, SDS-PAGE de la proteína total del lisado celular HEK293T usando geles de acrilamida al 10% según Ahn y otros (A) y Laemmli (B). Los geles teñidos con Coomassie® muestran alrededor de 20 bandas prominentes de proteínas con diferentes distribuciones de tamaño para ambos tipos de gel. Los patrones de migración de las proteínas fueron diferentes para ambos tipos de gel. Las proteínas con una masa molecular superior a 70 kD aparecen comprimidas en los geles de Tris-Glicina Laemmli, mientras que las proteínas marcadoras se distribuyeron linealmente en una amplia gama de tamaños utilizando un único gel de Ahn). Sin embargo, los patrones de migración de las proteínas difieren entre los geles individuales de Laemmli Tris-Glicina y Ahn en diferentes rangos de peso molecular. Para proteínas de 15 - 70 kilos Dalton (kD) el 10% de los geles Laemmli muestran la resolución más alta, mientras que el 10% de los geles Ahn muestran un patrón de migración lineal para tamaños de proteínas que van de 25 - 130 kD. Cuando se trata de separar una amplia gama de tamaños de proteínas en el mismo gel, existen dos opciones: Los geles Ahn simples parecen particularmente adecuados para proteínas de peso molecular medio y alto. Los geles Laemmli Tris-Glicina son mejores para proteínas pequeñas y medianas cuando se usan con la misma concentración de polímero de acrilamida.
2. Estabilidad
También estudiamos la estabilidad de las soluciones de acrilamida según Ahn y otros, almacenándolas durante un período de hasta seis meses a 2-8°C. Los geles de poliacrilamida hechos de tales soluciones no mostraron ningún efecto significativo sobre la calidad, incluso después de un almacenamiento prolongado. Sin embargo, los geles fundidos deben utilizarse en pocos días, ya que el valor del pH dentro del gel cambia rápidamente. Esto es válido tanto para los geles Laemmli Tris-Glicina como para los geles simples Ahn.
3. Western Blotting
Para la transferencia de proteínas el rendimiento varía dependiendo de la masa molecular de las proteínas (Fig. 2, Western blot de la proteína total de las células HEK293T después de SDS-PAGE según Ahn et Al. (A) y Laemmli (B). Se detectaron dos proteínas diferentes con anticuerpos específicos: Tubulina y una proteína de 25 kD. La expresión de la tubulina fue la misma en todas las células. La degradación de la proteína celular (o proteasómica) de la proteína marcada con su nombre fue inducida (+) o no inducida (-) en los experimentos. La tubulina se transfirió más eficientemente de los geles Ahn a las membranas de PVDF que de los geles Laemmli Tris-Glicina, mientras que la transferencia de la proteína 25 kD marcada con su etiqueta fue menos eficiente de los geles simples Ahn equivalentes.) La tubulina, una proteína de 66 kD, se transfirió de los geles Laemmli con un 55 ± 4 % menos de eficacia que los geles simples Ahn. En cambio, la transferencia de una proteína his-tag de 25 kD fue menos eficaz a partir de los geles Ahn en un 32 ± 12 % (n=4). En general, la transferencia de pequeñas proteínas es mejor usando los geles de Tris-Glicina de Laemmli, mientras que los geles simples de Ahn funcionaron mejor para las proteínas de mayor masa molecular.
4. Manejo
IA los usuarios entrevistados les gustó el procedimiento rápido y conveniente según Ahn et Al. El protocolo es fácilmente adoptado por los laboratorios que ya utilizan el protocolo Laemmli "clásico", ya que todos los aparatos, tampones y soluciones son los mismos para ambos procedimientos SDS-PAGE. Otros autores más escépticos también evaluaron el sistema de gel simple de Ahn y finalmente confirmaron el buen desempeño. Según G. Fritz (de la Univ. de Zurich, en Rehm 2007), las ventajas adicionales de los geles Ahn son: 1) Funcionan muy bien (no tienden a producir el efecto "sonrisas"). 2) Los geles funcionan a concentraciones más altas de poliacrilamida (hasta un 18 %) y 3) Evitan el gel viscoso de apilamiento.
Perspectivas
En la investigación, nuevos métodos son constantemente inventados creando nuevas perspectivas. Algunos de los métodos como la electroforesis en gel resisten la prueba del tiempo. Los métodos clásicos se mejoraron, se ajustaron y se añadieron nuevas características a lo largo de los años. ITW Reagents siempre se esfuerza por explorar protocolos de aplicación nuevos o mejorados para compartir productos prometedores con los clientes.
Bibliografía
- Ahn, T. et al. (2001) Anal. Biochem. 291, 300-303
- Davies, B.J. (1964) Ann. N. Y. Acad. Sci. 121, 404-427
- Eley et al. (1979) Anal. Biochem. 92, 411-419
- Fazekas de St. Groth, S. et al. (1963) Biochim. Biophys. Acta 71, 377-391
- Gordon, A.H. et al. (1950) Coll. Czech. Chem. Com. 15, 1
- Grabar, P. & Williams, C.A. (1953) Biochim. Biophys. Acta 10, 193-194
- Hjertén, S. (1961) Biochim. Biophys. Acta 53, 514-517
- Hjertén, S. (1967) Chromatogr. Rev. 9, 122-219
- Hjertén, S. (1983) J. Chromatogr. 270, 1-6
- Jorgenson, J.W. & Lukacs, K.D. (1981) Anal. Chem. 53, 1298
- Kendall, J. & Crittenden, F.D. (1923) Proc. Natl. Acad. Sci. USA 9, 75
- von Klobusitzky, D. & König, P. (1939) Arch. Exptl. Pathol. Pharmakol. Naunyn-Schmiedeberg 192, 271
- Kohlrausch, F. (1897) Wiedemanns Ann. (Ann. Phys. Chem.) 62, 209
- Kohn, J. (1957) Nature 180, 986
- Laemmli, U.K. (1970) Nature 227, 680-685
- Martin, A.J.P. (1942) unpublished results
- Merrill, R.C. et al. (1979) Proc. Natl. Acad. Sci. USA 76, 4335
- O’Farrell, P.H. (1975) J. Biol. Chem. 250, 4007-4021
- Ornstein, L. (1964) Ann. N. Y. Acad. Sci. 121, 321-349
- Panyim, S. & Chalkley, R. (1969) Arch. Biochem. Biophys. 130, 337-346
- Poduslo, J.F. (1981) Anal. Biochem. 114, 131
- Raymond, S. & Weintraub, L. (1959) Science 130, 711
- Reuss, F.F. (1809) Memoires de la Société Imperiale de Naturalistes de Moskou 2, 327
- Sanger, F. & Coulson, A.R. (1975) J. Mol. Biol. 94, 441-448
- Schägger, H. & von Jagow, G. (1987) Anal. Biochem. 166, 368-379
- Schägger, H. & von Jagow, G. (1991) Anal. Biochem. 199, 223-231
- Schwartz, D.C. & Cantor, C.R. (1984) Cell 37, 67-75
- Shapiro, A.L. et al. (1967) Biochim. Biophys. Res. Commun. 28, 815-820
- Smithies, O. (1955) Biochem. J. 61, 629-641
- Svensson, H. (1961) Acta Chem. Scand. 15, 325-341
- Tiselius, A. (1930) Nova Acta Regiae Societatis Scientarum Upsaliensis Ser IV, Vol. 7, No. 4
- Tiselius, A. (1937) Trans. Faraday Soc. 33, 524-531
- Towbin et al. (1979) Proc. Natl. Acad. Sci. USA 76, 4350–4354
- Vesterberg, O. (1969) Acta Chem. Scand. 23, 2653
- Weber, K & Osborn, M. (1969) J. Biol. Chem. 244, 4406
Revisiones: - Everaerts, F.M., Becker, J.L., & Verheggen, T.P.E.M. (1976) “Isotachophoresis: Theory, Instrumentation, and Applications”; Elsevier, Amsterdam
- Michov, B. (1995) „Elektrophorese: Theorie und Praxis“; Walter de Gruyter & Co, Berlin
- Vesterberg, O. (1993) “A short history of electrophoretic methods”; Electrophoresis, 14, 1243-1249
- Righetti, P.G. (2005) “Electrophoresis: The march of pennies, the march of dimes”; Journal of Chromatography A, 1079, 24-40
- Westermeier, R. (2004) “Electrophoresis in Practice: A Guide to Methods and Applications of DNA and Protein Separations”; 4th ed. Wiley-VCH, Weinheim