
In the center of molecular biology is one species of molecules: DNA.
DNA molecules are amplified and introduced into organisms by transformation or transfection, separated, stained, examined under the microscope, manipulated, sequenced and so on.
For all these techniques the initial step is to isolate DNA from the origin of interest.
This page provides you an overview of the different methods for nucleic acid isolation and offers a number of must-have reagents to obtain pure DNA and/or RNA from various sources.
Introduction
What does it mean technically when we talk about DNA extraction, isolation or purification - terms often used synonymously for getting preferably pure DNA from a sample? What are the mechanisms behind? How is it possible to separate distinct species of DNA?
On this page we focus on the isolation of the two DNA species that are mainly isolated in molecular biology labs – genomic DNA and bacterial plasmids. We give an overview about established DNA isolation techniques, their chemical background and we discuss their respective advantages and limitations.
Regarding the basic procedure, DNA extraction is simple and can be done using domestic products. Basically, all you require is a rich source of DNA, salt, water, dishwashing detergent, a coffee filter, high-proof alcohol and a stick to spool the precipitated DNA salt out of solution. For higher demands (regarding quantity and quality), of course, the method requires further refinement.
Purity and integrity of the DNA will affect the results of all subsequent applications, so highest quality of DNA is desirable for diagnosis and research.
Not only at home, but also in the lab, the isolation process steps are straightforward: First: destroy the cells and bring DNA into solution. Second: remove impurities by non-affinity (DNA remains in solution while everything else is removed) or affinity (DNA selectively binds to a solid matrix) based techniques.
It might be necessary to initially dissociate cells from the sample material (e.g. in case of tissue, bones, and plants). This procedure varies from sample to sample and uses almost every physical method that is finally resulting in a mash of DNA-containing material, including cutting, enzymatic treatment, mixing (using glass beads for example), grinding and powdering in a mortar or freezing in liquid nitrogen.
History of the discovery of DNA
The very first DNA isolation was described by the Swiss physician Friedrich Miescher in 1869. In the nucleus of leucocytes, obtained from the pus on surgical bandages, he identified an acid-soluble phosphorus and nitrogen containing substance which he called “nuclein”. For the isolation of this new substance, 1889 renamed into “nucleic acid” by his student Richard Altmann, Miescher used a successive solvent and alkaline extraction method with subsequent acidification. 1944, almost 50 years later, the better known story of DNA started with Avery, MacLeod and McCarty showing that the hereditary substance is in fact nucleic acids (and not protein as assumed in earlier studies). Followed by the observations of Erwin Chargaff (fixed ratios of purine and pyrimidine bases within a species) in 1949 and the X-ray studies of Rosalind Franklin and Maurice Wilkins in 1953 (helical structure), James Watson and Francis Crick finally discovered the molecular double helical structure of DNA created by the base pairing of adenine with thymine and cytosine with guanine in 1953.
Lysis
(see our page enzymes for nucleic acid biochemistry)
Even if DNA molecules can literally be found in the streets, the DNA of interest is typically enclosed in a cell. To gain the DNA, a process called lysis (Greek λύσις, lýsis from lýein "to separate") is required: cell wall (if present) and membrane are broken off, allowing everything formerly locked in the cell to be released into solution: DNA, RNA, proteins, lipids, and metabolites.
Lysis buffers for genomic DNA commonly include a detergent, sodium chloride, EDTA and enzymes to degrade proteins and RNA.
Ionic detergents such as SDS strongly destabilize the lipid bilayer of the cell membrane. Solubilizing the membrane lipids, they finally force the breakup of the cellular structure and support precipitation of lipids and proteins. EDTA assists destabilization of the cell wall, but its main task is to inhibit DNase activity by chelating divalent cations such as Mg2+. When it comes to protein degradation, proteinase K is the most common protease directly employed in the lysis solution. Conditions that promote enzyme inactivation like detergents, chaotropic salts, high temperature and changes in pH are well tolerated by proteinase K. Beside its high stability, proteinase K is characterized by a large number of cleavage sites and therefore perfectly suited to remove cellular and nuclear proteins that are attached to the DNA. Also RNA is degraded enzymatically. The ribonuclease RNase A, which hydrolyses RNA molecules into single nucleotides is heat stable as well, and, like Proteinase K it has no need for cofactors that might be complexed by EDTA.
In contrast, other enzymes such as lysozyme do not withstand the lysis conditions. Lysozyme effectively digests the bacterial cell wall (composed of peptidoglycane), but the enzyme is inhibited by surface-active agents like SDS. This is why for isolation of DNA from (gram-positive) bacteria the lysozyme treatment is performed prior to lysis.
David versus Goliath: Plasmids recover first!
Regarding prokaryotes and yeasts, plasmid isolations are at least as important as isolation of genomic DNA. Although they consist of the same building blocks and they both are double stranded, these two types of DNA can be clearly separated during the isolation procedure. Low molecular weight plasmids are easily purified from high molecular weight genomic DNA based on their solubility and re-annealing rate.
The preferred method for isolation of plasmid DNA is the alkaline lysis, a technique invented in 1979 (Birnboim & Doly, 1979). It works perfectly for conventional plasmids of a size of less than 15 kbp. The bacterial cell pellet is resuspended in Tris buffer with EDTA and RNase A. The lysis buffer, containing NaOH and SDS, is added to the homogenized cells and the solutions are mixed by gentle inversion. For isolation of plasmid DNA, the presence of chaotropic salts during lysis is counterproductive and will prevent the separation of the plasmid from the chromosome. So, in contrast to lysis buffer for genomic DNA, no chaotropic salts should be added to the alkaline lysis solution. Also, extensive incubation times in the lysis solution should be avoided, since they bear the risk of irreversible denaturation of the plasmid DNA.
Incubation of cells under alkaline conditions leads to denaturation of both DNA species, plasmids and genomic DNA.
During the lysis, as a consequence of the solubilisation of macromolecules, the solution gets clearer and highly viscous.
By adding a potassium acetate / acetic acid solution, the mixture is neutralized and all macromolecules except for plasmid DNA are precipitated. This is achieved as the small circular plasmid DNA quickly renatures. In contrast, the longer, tangled genomic DNA strands stay separated, sticking together with denatured proteins and coated with SDS molecules. The potassium ions in the neutralization buffer replace the SDS’s sodium ions and lead to the formation of a white, cloudy precipitate (Ish-Horowicz & Burke 1981) that is, together with residual cell debris, sedimented by centrifugation. The renatured plasmid DNA, unaffected by the large number of intermolecular interactions, remains in the supernatant and is further purified.
The crucial point in the separation process is to avoid vigorous mixing or vortexing during lysis and neutralization since genomic DNA easily breaks into small fragments that will finally contaminate the plasmid containing supernatant. DNA is very resistant against chemical influences, but, the longer the chain, the more sensitive is the DNA molecule to physical forces. It is inevitable that the large DNA chromosomes break during the purification process, but as long as they are not fragmented into small pieces this breakage is not of disadvantage. Even the opposite is the case: subsequent PCR reactions are more efficient due to the better melting of the broken DNA.
The final cleaning of the plasmid solution can be carried out using the same methods as for genomic DNA, e.g. by phenol-chloroform extraction, ethanol precipitation or using a silica column. The latter method is the most popular today, because it is very fast and provides high quality plasmid DNA, suitable for transformation, enzymatic digestion, mutagenesis etc.
Methods of DNA Isolation
When isolating DNA, the aim is of course to achieve maximum purity and quantity in a minimum of time with a minimum of costs. Unfortunately achieving all of this is rarely possible. Different methods of DNA isolation are employed, depending on sources, type of DNA (genomic, plasmid or organelles, e.g. mitochondrion), sample size, sample age and required purity. But not every method is suitable for every downstream process. The most important DNA isolation techniques are commercial solid-phase based kits (columns with silica or anion exchange matrices), phenol/chloroform extraction and monophasic reagents.
The traditional method: Phenol-chloroform Extraction
First described in 1956, the purification of nucleic acids using phenol to remove hydrophobic impurities is still widely employed today.
Phenol-chloroform extraction is cheap but effective. Followed by ethanol precipitation to remove salt and organic solvent impurities, the method yields high quantities of pure DNA. On the other hand, the procedure is time consuming, requires multiple tube transfers that bear the risk of contaminations (by foreign nucleic acids but also by phenol/chloroform carry-over) and involves hazardous chemicals. But still, the phenol extraction is often the method of choice for very small as well as large DNA fragments, and for old, partially degraded DNA.

Phenol as a cleaning agent for DNA
Based on the discovery that phenol is suitable to extract proteins from aqueous solutions (Grassmann & Deffner, 1953), Kirby reported phenol as an agent for nucleic acid purification (Kirby, 1956). Using a phenol-water mixture he extracted a tissue homogenate and showed that RNA was separated in the aqueous phase, while proteins associated with DNA were transferred to the interphase. Shortly after, Kirby found, that in the presence of certain anionic salts (e.g. p-aminosalicylate and sodium benzoate) both nucleic acid species, DNA and RNA, are enriched in the aqueous phase (Kirby, 1957). Today, Kirby’s method is still up-to-date, apart from a few adaptions (anionic detergents like SDS replaced the anionic salts, addition of chloroform and isoamyl alcohol, and equilibration of the pH).
Polar, hydrophilic compounds like DNA, RNA and proteins commonly dissolve best in polar solvents (with water as the solvent offering maximum polarity). But in contrast to nucleic acids, proteins provide a number of non-polar structures as well. The non-polar side chains of phenylalanine, leucine, isoleucine, valine, proline, methionine and alanine enable the protein to stay in solution when exposed to a less polar or even non-polar solvent. The proteins rearrange exposing the non-polar side chains to the surface, while the charged and polar residues become buried inside the protein complex. These features enable the extraction of proteins out of an aqueous phase by a less or even non-polar solvent. Phenol is clearly less polar than water despite its electronegative oxygen atom; because the phenyl ring renders the electron density spread all over the molecule but not concentrated on the oxygen atom.
For DNA isolation, the phenol has to be pH-equilibrated with tris to a final pH of >7.8 to ensure that the DNA is negatively charged and therefore insoluble in the organic phase. Starting with the cell lysate, an equal volume of tris-buffered phenol-chloroform (1:1), or tris-buffered phenol-chloroform-isoamyl alcohol (25:24:1) is added and the solution is mixed by vortexing (small DNA molecules of <10 kb), gently shaking (10-30 kb) or slowly inverting or rotating (>30 kb). Chloroform efficiently denatures proteins, avoids the retention of water in the organic phase and improves the phase separation by increasing the density of the organic phase. The addition of a small volume of isoamyl alcohol reduces foaming during the extraction process, and, aiming at RNA isolation, guarantees the deactivation of RNases (Green & Sambrook, 2012).
Centrifugation accelerates the separation of the two phases, resulting in the aqueous phase with the lower specific gravity on top. But caution: A high salt content or high amounts of sucrose in the aqueous solution can result in an inversion of the two phases! So it is highly recommended to make sure the right solution is further processed. Since equilibrated phenol commonly contains 8-hydroxyquinoline as a stabilizer, the organic phase can be identified by its yellow color. After organic extraction, DNA (and RNA, if no RNase A has been added during lysis) is still in the aqueous phase while denatured proteins have moved into the interphase and lipids have been transferred to the organic phase.
After repeated extractions with the phenol-containing solvent, the DNA-containing aqueous phase is extracted a few times with chloroform (or chloroform/isoamyl alcohol), to remove residual phenol. Finally, the pure DNA is precipitated from solution using ethanol or isopropanol.
PanReac AppliChem's products for Phenol-Chloroform Extraction of DNA
| Prod. No. | Description | Comment |
| A1153 | Phenol equilibrated, stabilized 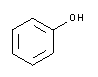 |
Pure phenol is a colorless and crystalline substance. Liquefied phenol is susceptible to oxidation (especially when the pH is equilibrated with Tris) and the phenolic oxidation products introduce strand breaks into nucleic acid molecules and promote cross-linking. Phenol solutions should be clear and colorless, pink or brownish solutions should be discarded. To prevent oxidation, 8-hydroxyquinoline is frequently added to liquefied phenol. The shelf life of equilibrated and 8-hydroxyquinoline-stabilized phenol is approximately 9 months. As a positive side effect, 8-hydroxyquinoline partially inhibits ribonucleases. |
| A1624 | Phenol water-saturated, stabilized | |
| A1578 | Phenol water-saturated, non-stabilized | |
| A1594 | Phenol crystalline | |
| A0889 | Phenol equilibrated, stabilized : Chloroform : Isoamyl alcohol 25:24:1 | |
| A3691 | Chloroform | |
| A2610 | Isoamyl alcohol |
The all in one solution: Acidic guanidinium thiocyanate-phenol extraction
Admittedly, this technique does not really aim at DNA isolation in the first place, but nevertheless it is possible to use it on this purpose. And since this technique offers interesting possibilities it should be mentioned in this article.
Invented by Chomczynski & Sacchi in 1987 for isolation of RNA, acidic guanidinium thiocyanate-phenol extraction allows cell lysis and successive separation of RNA, DNA and proteins using one reagent. The technique combines the effect of chaotropic salts on structure and solubility of macromolecules with the extraction properties of acidic phenol. Commercialized under names like Trizol®, TRI REAGENT® and TRItidy™ G, Chomczynski’s reagent conquered the laboratories and still belongs to the number one choice for RNA isolation.
The birth of monophasic reagents
The history of acidic guanidinium thiocyanate - phenol extraction (or the “single-step” method) invented by Chomczynski & Sacchi is in first place the history of RNA isolation. In 1951, the usage of guanidinium chloride in RNA isolation was firstly described (Volkin & Carter, 1951). After Kirby’s introduction of phenol as a deproteinization reagent, guanidinium salts temporarily seem to lose their importance, but “guanidinium chloride in the isolation of nucleic acids” was focused again in the 1960s (Cox 1968). In 1979, Chirgwin et al. published a method to isolate undegraded RNA from ribonuclease-rich tissues employing guanidinium thiocyanate instead of guanidinium chloride. In 1987, Chomczynski & Sacchi initially used a combination of guanidinium thiocyanate and phenol-chloroform extraction for RNA isolation. Based on the same principle, the method was expanded for simultaneous isolation of RNA, DNA and proteins (Chomczynski, 1993). Two years later, further modifications were published employing bromochloropropane (Chomczynski & Mackey, 1995a) which is less toxic than chloroform and forms a tighter interphase, and isopropanol (Chomczynski & Mackey, 1995b) for improved isolation of RNA from polysaccharide- and proteoglycan-rich sources. The development finally also focused on the isolation of genomic DNA (Chomczynski et al., 1997).
The monophasic solution contains water, phenol, guanidinium thiocyanate (also often referred to as guanidine thiocyanate), β-mercaptoethanol and a detergent. The chaotropic salt guanidinium thiocyanate lyses the cells, denatures the released macromolecules and inactivates RNases and other enzymes. As a detergent, lauroylsarcosine (“sarkosyl”) is a good choice, since, in contrast to SDS, lauroylsarcosine shows high solubility in chaotropic high salt buffers. Addition of sarkosyl to cell or tissue homogenates improves the purity of the RNA isolated by guanidinium salts and reduces foaming during homogenisation (MacDonald et al., 1987).

After lysis of the cell in the reagent (e.g. by repetitive pipetting), chloroform (or alternatively bromochloropropane) is added, leading to the generation of a second phase. While DNA and proteins enrich in the newly formed organic phase and the interphase, RNA is selectively retained in the aqueous phase. RNA, DNA and proteins are isolated by alcohol precipitation.
Why is hydrophilic DNA transferred into the organic phase? And how is it possible that RNA, in contrast, is not extracted by the phenol-chloroform mixture? DNA and RNA seem to be very similar at first sight and, under neutral conditions (pH 7-8) both molecules remain in the aqueous phase as expected. However, usage of non-equilibrated, and therefore acidic phenol solution, enable the enrichment of DNA molecules in the non-polar organic phase. So why do these two types of nucleic acids behave differently under acidic conditions? The answer mainly lies in the structural differences between DNA and RNA: at the prevailing pH of 4-5, the phosphate groups of the doubled stranded (and less acidic) DNA are protonated and the affinity of the now non-charged (but still double stranded) DNA molecules to the organic solvent strongly increases. The phosphate backbone of the single stranded RNA is largely protonated as well, but due to the exposure of the purine and pyrimidine bases, the RNA is able to form hydrogen bonds with the surrounding water molecules (Zumbo, 2011). As a result, RNA does not lose its hydrophilic properties and still prefers the aqueous phase.
| PanReac AppliChem's monophasic reagents: acidic guanidinium thiocyanate-phenol extraction | ||
| Prod. No. | Description | Comment |
| A4051 | TRItidy G™ | Ready-to-use solution for sequential isolation of RNA, DNA and proteins. |
| A3418 | DNA - Isolation reagent for genomic DNA | Phenol-free ready-to-use reagent for the isolation of genomic DNA from human, animal (incl. mouse tail), plant, yeast, bacterial and viral origin. |
Fast desalting: Ethanol precipitation
Today, ethanol precipitation is often used as the final step in DNA isolation by organic extraction or anion-exchange chromatography. The possibility to precipitate DNA out of solution enables not only removal of alcohol soluble salts, residual organic solvents and detergents; it also offers an opportunity to concentrate DNA.
In neutral aqueous solutions, the negatively charged phosphate backbone of DNA molecules is saturated with the highly polar water molecules that prevent interaction with cationic molecules. Increasing concentrations of ethanol lead to a disruption of the hydrate shell and allow the formation of ionic bonds between the phosphate groups and positively charged ions. As a consequence, in solutions containing at least 65% ethanol and a sufficient amount of cations, the previously negatively charged DNA molecules are neutralized. The loss of charge minimizes the repulsive forces between the molecules and finally makes DNA to precipitate. The accurate salt concentration is crucial to ensure that on the one hand, all DNA is recovered from solution, and on the other hand, the anionic salts do not co-precipitate. To improve the efficiency, the target nucleic acids often are co-precipitated with nuclease-free inert “carriers” such as glycogen or linear polyacrylamide.
The most common salts used in ethanol precipitations are ammonium acetate, sodium acetate and sodium chloride.
A sodium acetate solution at acidic pH of 5.2 is the standard reagent for nucleic acid precipitation. Sodium chloride is the first choice if SDS is included in the sample ensuring its solubility in the presence of alcohol and enabling the precipitation of detergent-free DNA. If the sample is contaminated by dNTPs or oligosaccharides (which includes DNA samples obtained from agarose gels by agarase digestion), ammonium acetate is most suitable since the ammonium cations prevent co-precipitation of these two species (source: Green & Sambrook, 2012).
Isopropanol can be used alternatively for DNA precipitation; it is mostly used for large volumes since only half of the quantity is required to dehydrate the DNA molecules. Unfortunately, the solubility of salts in solutions with 35% isopropanol is reduced compared to 65% ethanol, increasing the risk of salt co-precipitations. Furthermore, it is less volatile than ethanol and therefore harder to remove, increasing the risk of alcohol carry-over into the final sample.
| PanReac AppliChem's products for alcohol precipitation of nucleic acids | |
| Prod. No. | Description |
| A3678 | Ethanol absolute 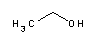 |
| A3928 | 2-Propanol 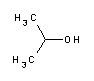 |
| A4555 | Sodium acetate anhydrous |
The convenient method: Commercial kits employing pure silica and anion exchange columns
Silica-based spin kits:
The ability of DNA to bind rapidly and selectively to silicates at high salt concentrations under alkaline conditions was first described in 1979 (Vogelstein & Gillespie, 1979): DNA was removed from agarose gels and bound to glass in the presence of sodium iodide.
Positively charged ions shield the acidic silica surface promoting adsorption of the DNA molecules through the negatively charged DNA backbone. The type of bridging cation determines the strength of binding (Romanowski et al., 1991). Washing with chaotropic salt solutions removes residual protein impurities without affecting the immobilized DNA. Not only salts, also addition of alcohol influences the interaction between matrix and DNA. The silica-bound DNA withstands washing procedures employing 70% ethanol solutions that displace the excess of salts, metabolites, RNA, carbohydrates, and other alcohol soluble biomolecules. Only elution with pure water or low salt solutions (e.g. TE buffer) releases the DNA. This is because the large excess of water molecules replaces the ionic bonds and rehydrates DNA as well as the surface of the silica particles.
The silica-based spin kits are very popular for isolation of genomic DNA as well as for isolation of plasmids. The purity of the eluted DNA is high, and due to the low salt elution conditions, further desalting of the DNA is not necessary. The method is fast, easy to perform, and provides reproducible quantities and quality. Unfortunately, the columns are not suitable for very small DNA fragments. The smaller the DNA molecules, the tighter the binding between silica matrix and DNA. As a consequence, small DNAs cannot effectively be recovered from the column (Green & Sambrook, 2012). A further drawback is that the binding capacity of silica is only moderate. Therefore, these “mini” columns are only suitable for small quantities of DNA, typically up to 20 µg.
The meaning of chaotropic salts in nucleic acid isolation processes.
Chaotropic agents like guanidinium salts, urea or lithium perchlorate show a number of positive effects on the isolation process:
- lysis of the cell membrane,
- denaturation of proteins, DNA and other macromolecules,
- inactivation of nucleases,
- promotion of nucleic acid binding to pure silica material.
All these features are based on the same mode of action. The chaotropic substances interfere with intracellular interactions based on hydrogen bonds, hydrophobic effects and van der Waals forces resulting in denaturation of proteins (as a consequence protein activity is significantly reduced) and DNA, but also in reorganization and finally collapse of the membrane. The accumulation of chaotropic salts in the hydrophobic region of the lipid bilayer strongly compromises membrane integrity. By replacing the existing hydrogen bonds with adjacent water molecules by salt bridges, the chaotropic salts destroy the hydration shell that is surrounding the nucleic acids, decrease their solubility, mask charges and mediate adsorption to silica surfaces.
Anion exchange-based spin kits
The principle of anion exchange totally differs from pure silica-based DNA purification. In contrast to the negatively charged silicate surface, the matrix material possesses a high density of positive charges. The hydrophilic anion exchange resin consists of large-pored silica beads coated with cationic groups. Maximum binding of DNA takes place under slightly acidic low-salt conditions that enable a direct and undisturbed ionic interaction of the DNA’s phosphate groups with the cationic surface of the resin.
Impurities are removed by washing with medium salt buffers (~ 0.8 - 1.0 M NaCl, depending on the pH). The DNA is eluted with high salt solutions at a pH around 8. The excess of anions replaces the bound DNA molecules and saturates the surface of the anion exchange matrix. Anion exchange materials guarantee a very pure DNA and, compared to pure silica, offer a strongly increased binding capacity due to its high charge density. Therefore, anion exchange matrices are preferentially used for large scale DNA isolations. Disadvantage are the elution conditions, namely the requirement of very high salt concentrations to release the DNA from the column. Subsequent desalting is often essential for downstream processes.
Columns of commercial spin kits are commonly discarded after a single use, since 5-10 % of the DNA remains in the matrix. Of course, this is a waste of resources and money. And it is not mandatory because the matrix itself is still working perfectly (Green & Sambrook, 2012). PanReac AppliChem offers a product for regeneration of pure silica and anion exchange columns that even improves the binding capacity. The column regeneration kits maxXbond™ are based on the ExitusPlus™ technology (see chapter about Decontamination), bio-degradable and non-hazardous.
PanReac AppliChem's column-based DNA and RNA Purification Spin-Kits [AX: Anion Exchanger; SI: Pure Silica]
| Prod. No. | Description | Comment |
| Purification of DNA and DNA fragments | ||
| A5193 | DNA Isolation Spin-Kit Agarose | Standard Gel Extraction Kit SI] for Isolation of DNA from agarose gels |
| Decontamination & Regeneration of spin-columns | ||
| MB007 | maxXbond™ | Silica matrices are a key technology for the purification of DNA. Today the rapid isolation of pure DNA samples is essential for a variety of molecular biology protocols in research and commercial applications. Products with silica matrices are of high quality and high value. Their major disadvantage is that they can only be used once because after elution substantial amounts of DNA remain attached to the silica matrix and the binding capacity is reduced. To solve this problem we developed the first regeneration system for silica matrices: maxXbond™ for pure silica and maxXbond™AX for anion exchange columns. Two innovative solutions remove all nucleic acids and extraneous material from the matrix and restore the original binding capacity. |
References
- Arena, A. (2010) Invest. Sci. J. 2(2): 20-29
- Birnboim, H.C. & Doly, J. (1979) Nucleic Acids Res 7: 1513-1523
- Chirgwin, J.M., Przybyla, A.E., MacDonald, R.J. and Rutter, W.R. (1979) Biochemistry 18: 5294-5299
- Chomczynski, P. & Sacchi, N. (1987) Anal. Biochem. 162: 156-159
- Chomczynski, P. (1993) BioTechniques 15: 532-537
- Chomczynski, P. & Mackey, K. (1995a) Anal. Biochem. 225: 163-164
- Chomczynski, P. & Mackey, K. (1995b) BioTechniques 19: 942-945
- Chomczynski, P., Mackey, K., Drews, R. and Wilfinger, W. (1997) BioTechniques 22(3): 550-3.
- Cox, R.A. (1968) in Methods in Enzymology (Grossman L & Moldave K., Eds.), 12-B: 120-129, Academic Press, Orlando, Florida
- Grassman W. & Deffner, G. (1953) Hoppe Zeylers Z Physiol Chem 293: 89-98.
- Green, M.R. & Sambrook, J. (2012) Molecular Cloning: A Laboratory Manual, Fourth Edition, Cold Spring Harbour Laboratory Press, Cold Spring Harbour, New York.
- Ish-Horowicz, D. & Burke, J.F. (1981) Nucleic Acids Res 9: 2989-2998
- Kirby, K.S. (1956) Biochem J 64:405-408
- Kirby, K.S. (1957) Biochem J 66:495-504
- MacDonald, R.J., Swift, G.H., Przybyla, A.E. and Chirgwin J.M. (1987) Methods Enzymol. 152: 219-227
- Romanowski, G., Lorenz, M.G. and Wackernagel, W. (1991) Appl Environ Microbiol 57: 1057-1061
- Vogelstein, B. & Gillespie, D. (1979) Proc. Natl. Acad. Sci. USA 76, 615-619
- Volkin, E. & Carter, C.F. (1951) J. Am. Chem. Soc. 73(4): 1516-1519
- Zumbo, P. (2011) White paper. physiology.med.cornell.edu/faculty/mason/lab/zumbo/files/rna_phenol_chloroform.pdf