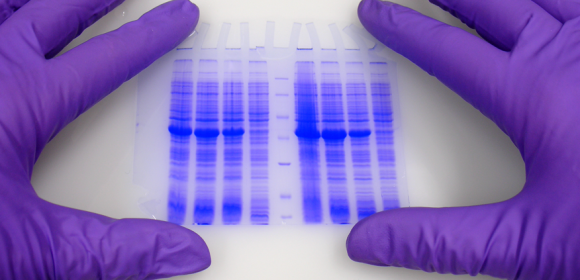

In den Biowissenschaften ist die Erforschung von Proteinen eines der wichtigsten Gebiete. Die Expression dessen, was in unseren Genen kodiert ist, ist nachweisbar und wird heute auch unter dem Begriff Proteomik zusammengefasst. In den letzten Jahren und Jahrzehnten wurden viele verschiedene Techniken entwickelt. Eine der bekanntesten und am häufigsten verwendeten ist die Polyacrylamid-Gel-Elektrophorese PAGE.
Polyacrylamide Gel Electrophoresis, PAGE
Polyacrylamid-Gel-Elektrophorese wird typischerweise zur Trennung von Proteinen verwendet, aber auch DNA (insbesondere zur Visualisierung von Unterschieden zwischen kleinen Fragmenten) kann durch PAGE getrennt werden. Die Matrix besteht aus Acrylamid-Strängen, die mit N,N-Methylenbisacrylamid vernetzt sind. ITW Reagents bietet Reagenzien und Fertigmischungen für SDS-PAGE, native PAGE, denaturierende und nicht-denaturierende DNA-PAGE sowie für die Sequenzierung von Gelen.
Acrylamidmischungen
Die Trennkapazität eines Polyacrylamidgels wird durch das Mischungsverhältnis von Acrylamid zu Bisacrylamid bestimmt. Je geringer das Verhältnis dieser beiden Komponenten im Gemisch, desto höher ist der Vernetzungsgrad. Das bedeutet, dass ein 6%iges Gel, hergestellt aus einer Stammlösung mit einem Mischungsverhältnis von 29 : 1, einen höheren Vernetzungsgrad aufweist als ein 6%iges Gel, hergestellt aus einer Stammlösung mit einem Mischungsverhältnis von 37,5 : 1.
Für die meisten Anwendungen wird ein Acrylamid: Bisacrylamid-Verhältnis von 29 : 1 oder 37,5 : 1 verwendet (zur elektrophoretischen Trennung von Nukleinsäuren oder Proteinen). Das Mischungsverhältnis von 19: 1 ist die Lösung der Wahl für die DNA-Sequenzierung. Die Herstellung wird vereinfacht, indem 30% oder 40% wässrige Acrylamid-Stammlösungen mit dem gewünschten Verhältnis verwendet werden.
Acrylamidlösungen werden oft als "gasstabilisiert" bezeichnet und bieten eine längere Haltbarkeit. Das "Gas" ist einfach Sauerstoff, was eine spontane Polymerisation von Acrylamidlösungen vermeiden soll. Da die Polymerisationsinitiatoren APS und TEMED jedoch im Übermaß zugesetzt werden, ist eine "Entgasung" (teilweise im Labor vor der Polymerisationsinitiierung) eigentlich unnötig.Die unerwünschte spontane Polymerisation wird durch Radikale ausgelöst, die typischerweise aus Acrylsäure stammen. Schlechtere Qualitäten enthalten oft nachweisbare Spuren von Acrylsäure und Lösungen auf Basis dieser Qualitäten bergen ein hohes Risiko, spontan zu polymerisieren. Im Gegensatz dazu sind die vierfach rekristallisierten Typen ("4K") jedoch frei von Acrylsäure. Für unsere Molekularbiologie-Qualität wird nur 4K-Acrylamid verwendet, das speziell auf das Fehlen von DNasen, RNasen und Proteasen getestet wurde!Bei niedrigen Temperaturen von 2-8°C wird der Sauerstoffaustausch innerhalb der Acrylamidlösung reduziert und die spontane Polymerisation erleichtert. Daher empfehlen wir die Lagerung bei Umgebungstemperatur. Die folgende Tabelle listet die gängigsten Acrylamidmischungen auf; weitere Acrylamidlösungen sind im Shop oder auf Anfrage erhältlich.
Verwendung von Acrylamidmischungen
Die Acrylamid-Stammlösungen werden verdünnt, um die gewünschte Monomerkonzentration zu erhalten. Das Gesamtvolumen der erforderlichen Menge an Gel-Lösung (z.B. 100 ml) wird durch den Gehalt der Lösung (z.B. 30 %) dividiert und mit der gewünschten Endkonzentration (z.B. 6 % Acrylamid) multipliziert, um das erforderliche Volumen der Stammlösung zu erhalten:
(100 ml / 30 %) * 6 % = 20 ml
Zur Herstellung von 100 ml eines 6 %igen Acrylamidgels werden 20 ml der 30 %igen Acrylamid-Stammlösung in die Gelmischung gegeben.
Selbstverständlich können Sie unsere Acrylamidmischungen auch in Pulverform anfordern. Aber Vorsicht, das Acrylamid-Monomer ist ein stark ansammelndes Neurotoxin! Daher sollten bei der Handhabung von kristallinem Acrylamid Handschuhe und eine Gesichtsmaske getragen werden.

Pufferlösungen, SDS, APS, APS, TEMED
Der am häufigsten verwendete Elektrophoresepuffer für SDS-PAGE ist SDS-tris-glycin, der sogenannte Laemmli-Puffer. Eine Alternative ist das von Schaegger & Jagow entwickelte tris-tricine-SDS-System. Für native Proteingele ist der Tris-Glycin-Puffer die erste Wahl.
Um die Polymerisation einzuleiten, werden 100 µl 10 % APS und 5-10 µl TEMED pro 10 ml Gel-Lösung zugegeben. Da die Polymerisation durch TEMED und den Radikalinitiator APS sehr schnell induziert wird, sollte das Gel sofort gegossen werden. Das Abkühlen der Lösung verzögert den Polymerisationsprozess. APS (Ammoniumpersulfat, A1142) ist in wässriger Lösung nicht sehr stabil (üblicherweise wird eine 10 %ige Stammlösung in Wasser hergestellt), aber erfahrungsgemäß kann die Lösung mehrere Wochen bei 2-8°C oder monatelang bei -20°C gelagert werden, ohne ihre Aktivität zu verlieren.
Pufferkomponenten und andere Chemikalien für die Polyacrylamid-Gel-Elektrophorese von Proteinen
| Prod. Nr. | Beschreibung | Kommentar |
|---|---|---|
| A1142 | Ammoniumperoxodisulfat | APS; die empfohlene Endkonzentration in Acrylamidgelen beträgt 0,1 % (w/v). Bereiten Sie eine 10 %ige (G/V) Stammlösung für eine einfache Anwendung vor. |
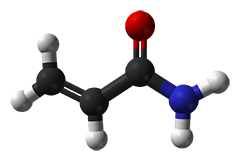
| A1148 | TEMED | N,N,N,N',N'-Tetramethylethylendiamin; die empfohlene Endkonzentration in Acrylamidgelen beträgt 0,1 % (v/v). |
| A0676 | SDS - 10%ige Lösung | Natriumdodecylsulfat; auch als 20 %ige Lösung (A0675) oder kristallin (e.g. A2572). |
Gebrauchsfertige Acrylamidlösungen für SDS-PAGE
Sparen Sie Zeit und sichern Sie sich reproduzierbare Ergebnisse: Unsere gebrauchsfertigen Gel-Lösungen für SDS-PAGE sind Ihr bevorzugtes Produkt für eine einfache und schnelle Gelpräparation. ITW Reagents bietet gebrauchsfertige Lösungen zum Stapeln und Lösen von Gelen nach Laemmli, basierend auf einem Acrylamid/Bisacrylamid-Verhältnis von 29 : 1 mit 0,1% SDS und Tris.
Zur Herstellung des Acrylamid-Gels werden 100 ml jeder gebrauchsfertigen Lösung (siehe Tabelle unten) mit 1 ml APS (aus einer 10%igen Stammlösung auf Basis von A1142) und 50 µl TEMED (A1148) ergänzt, um die Polymerisation des Gels einzuleiten. Für die Elektrophorese wird ein SDS-Tris-Glycin-Puffer, A1415) verwendet.
Proteinmarker und Proteinfarbstoffe
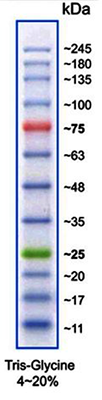
(Bild: Protein Marker VI)
Proteingrößenstandards für SDS-PAGE
| Prod. Nr. | Beschreibung | Kommentar |
| A8889 | Protein Marker VI (10-245) vorgefärbt | 12 Banden; gebrauchsfertiger und vordefinierter Marker für eine bessere Erkennung und Orientierung |
Für die unspezifische Färbung der Proteinbanden auf dem Polyacrylamidgel wird hauptsächlich Coomassie® Brilliant Blue R-250 verwendet. Wenn eine höhere Empfindlichkeit erforderlich ist, wird eine Silbernitratfärbung eingesetzt. Vor oder während des Färbevorgangs müssen die Proteine in der Gelmatrix fixiert werden. Dies geschieht durch eine wässrige Mischung aus Ethanol und Essigsäure oder Trichloressigsäure, die zur Denaturierung und Fällung von Proteinen führt. Für die Fixierung von kleinen basischen Proteinen ist Formaldehyd besser geeignet.
Proteinfarbstoffe zum Färben von Polyacrylamidgelen
| Prod. Nr. | Beschreibung | Kommentar |
| A1092 | Coomassie® Brilliantblau R-250 | Eines der am häufigsten verwendeten Färbemittel für Proteine in SDS-PAGE. Der Protein-Farbstoff-Komplex hat ein Absorptionsmaximum bei 549 nm. Pro positiv geladener Aminosäure werden ca. 1,5 - 3 Moleküle von Coomassie® Brilliant Blue R-250 gebunden. Die Empfindlichkeit liegt bei etwa 200-400 ng Protein/Band. |
| A3480 | Coomassie® Brilliantblau G-250 | Die Hauptanwendung dieses Farbstoffs ist der Bradford-Assay (Bestimmung der Proteinkonzentration), aber auch die Färbung von Proteinen in Polyacrylamidgelen ist möglich. |
| A3930 | Rhodamin B  |
Siehe Eriochromschwarz T (131439). |

Die Geschichte der Elektrophorese - Vergangenheit und Gegenwart
Geladene Moleküle wandern in einem elektrischen Feld. Dies ist das Grundprinzip der Elektrophorese, eine Methode, die heute in fast jedem Biologielabor eingesetzt wird und die für die meisten Trenntechniken und Analysemethoden grundlegend ist.
Wenn wir den Begriff Elektrophorese hören, werden wir wahrscheinlich an Gelelektrophorese, Agarose oder Acrylamid denken, insbesondere an SDS-PAGE - die heute die beliebteste und am weitesten verbreitete elektrophoretische Methode in der Forschung weltweit ist. Aber das Feld der elektrophoretischen Methoden und Anwendungen ist sehr breit gefächert und im Gegensatz zu den heute dominierenden Techniken wurden die ersten Schritte der Elektrophorese in freier Lösung ohne jegliche Trägermedien durchgeführt.
Die Anfangsjahre
Wie begann diese Erfolgsgeschichte? Die grundlegende Theorie der Elektrophorese wurde vor mehr als 200 Jahren entwickelt, aber erst in den 1930er Jahren präsentierte Tiselius die so genannte Bewegungsgrenzenelektrophorese. Diese Technik der Elektrophorese mit freier Lösung war geeignet, die Mobilität geladener Moleküle zu untersuchen. Im "Tiselius-Apparat" wurden die durch elektrophoretisch migrierende Proteine gebildeten "beweglichen Grenzen" durch die Veränderungen der Lichtabsorption oder des Brechungsindex gemessen. Im Gegensatz zu den heutigen Methoden wurde nie eine vollständige Trennung der Komponenten eines Gemisches erreicht, unabhängig davon, wie lange das Experiment dauerte. Nur eine Teilanalyse der schnellsten und langsamsten Migrationsverbindungen war möglich.
In den 1950er Jahren wurde die Bewegungsgrenzenelektrophorese durch die Zonenelektrophorese übertroffen, ein anderes Prinzip der Elektrophorese, das erstmals 1939 beschrieben wurde. In der Zonenelektrophorese wurden Proteine, Nukleoside, Aminosäuren und andere Moleküle mit Hilfe eines Trägermediums physikalisch voneinander getrennt. Anfangs wurden Filterpapiere verwendet, bald wurde das Verfahren aber durch alternative Träger wie Celluloseacetat (Kohn 1957), Stärke (Smithies 1955), Polyacrylamid (Raymond & Weintraub 1959) und Agarose (Hjertén 1961) stark verbessert. Das Trägermedium verhindert eine Sedimentation der Moleküle und ermöglicht - im Gegensatz zur Bewegungsgrenzenelektrophorese - eine vollständige Trennung.
Die Erfindung von PAGE
Mit den 1960er Jahren begann eine neue Ära auf dem Gebiet der Elektrophorese. Neue Techniken wurden eingeführt, bereits bestehende Methoden weitgehend verbessert. Die Disk-Elektrophorese wurde ebenso entwickelt wie die isoelektrische Fokussierung. Coomassie® Brilliantblau wurde zunächst als Farbstoff verwendet, um Proteinbanden nach der Gelelektrophorese zu visualisieren, und SDS wurde identifiziert, um die Nettoladung von Proteinen während der Polyacrylamid-Elektrophorese zu "überdecken".
Fünf Jahre nach der Einführung von Polyacrylamidgelen für die Protein-Elektrophorese durch Raymond & Weintraub wurde dieses Material von Ornstein und Davis als Unterstützung für eine Technik verwendet, die bisher in fast jedem biochemischen Labor bekannt und angewendet wird: 1964 entwickelten sie die Disk-Elektrophorese, welche die Zonenelektrophorese mit der Isotachophorese kombiniert.
Die Isotachophorese wird in einem diskontinuierlichen Puffersystem durchgeführt.
Durch Isotachophorese - zunächst als Ionenmigrationsverfahren (Kendall & Crittenden 1923) oder Verdrängungselektrophorese (Martin 1942) bezeichnet - können scharfe Grenzen zwischen den Probenbestandteilen erzeugt werden. Im Gegensatz zur Zonenelektrophorese wird die Isotachophorese in einem diskontinuierlichen Puffersystem durchgeführt, das aus einem führenden Elektrolyt mit hoher Mobilität und einem terminierenden oder schleppenden Ion mit niedriger Mobilität besteht. Nach dem Aufbau des elektrischen Feldes migrieren die Moleküle mit unterschiedlicher Geschwindigkeit und bilden völlig getrennt voneinander Stapel; das Molekül mit der höchsten Mobilität folgt direkt dem Leition, Moleküle mit der geringsten Mobilität wandern direkt vor dem terminierenden Elektrolyten. Bisher nichts Besonderes. Je schneller sich die Ionen bewegen, desto geringer ist das elektrische Umgebungsfeld, während die langsameren ein höheres Feld erzeugen. Infolgedessen migrieren nach der Trennung alle Moleküle mit der gleichen Geschwindigkeit (und das bedeutet der Name Isotachophorese wörtlich: Migration mit gleicher Geschwindigkeit): Die zunächst schneller migrierenden Ionen werden durch das umgebende schwächere Feld verzögert; die langsameren Ionen werden durch das stärkere Feld um sie herum beschleunigt. Aus diesem Grund hat das System einen Selbstschärfeffekt; sobald ein Ion aus seinem eigenen Band diffundiert, wird es entweder entschleunigt oder beschleunigt und so rückwärts in sein ursprüngliches Band gedrückt.
Wieder zurück zum Prinzip der Disk-Elektrophorese. Durch die Verwendung von zwei verschiedenen Separationsbereichen (Stapelung und Auflösung des Gels) und einem diskontinuierlichen Puffersystem verhinderten Ornstein und Davis die Bildung von Proteinaggregaten beim Eintritt in die Gelmatrix und unterstützten die Trennung in klar definierte Banden.
Das Stapelgel zeichnet sich durch einen niedrigeren pH-Wert und größere Poren als das angrenzende Lösungsgel aus.
Das Stapelgel zeichnet sich durch einen niedrigeren pH-Wert und größere Poren als das angrenzende Lösungsgel aus. Beide Gele enthalten nur Chloridionen (mit Tris als Gegenion), während der Elektrodenpuffer nur Glycin enthält. Zunächst werden die Proteine "gestapelt" und nach dem Prinzip der Isotachophorese konzentriert. Aufgrund der großen Poren des Stapelgels hat die Größe des Moleküls keinen Einfluss auf seine Mobilität. Die Nettoladung von Glycin ist beim pH-Wert des Stapelgels nahezu Null, daher fungiert Glycin als terminierendes Ion. Wenn die Proteinfront die Grenze zum engmaschigen Lösungsgel erreicht, passieren die kleinen Glycinmoleküle die Proteine, gelangen in den Lösungsbereich, werden höher geladen und bewegen sich zusammen mit den Chloridionen vor der Proteinfraktion. Sobald die Proteine von dem homogenen Puffer umgeben sind, beginnen sie sich nach den Prinzipien der Zonenelektrophorese zu trennen: Ihre Mobilität hängt nun von ihrer Ladung und Größe ab, was schließlich zu einer Neuordnung der Proteinrangfolge führt.
Als Laemmli 1970 seine berühmte Arbeit über die T4-Phagenproteintrennung veröffentlichte, verwendete er Ornstein und Davis' Tris-Glycin-Chlorid-Puffersystem. Aber anstatt eine "native" PAGE zu führen, fügte er SDS hinzu, das 1967 von Shapiro et al. eingeführt wurde. Bei SDS-PAGE basiert der Trennprozess nicht mehr auf der spezifischen Nettoladung der Proteine, sondern auf deren unterschiedlichen Molekulargewichten. Die Kombination von SDS-PAGE mit "Laemmli"-Puffer ist bis heute die am häufigsten verwendete Technik zur Trennung von Proteinen.
Ein weiterer Meilenstein in der Entwicklung der Gelelektrophorese war die isoelektrische Trennung von Proteinen. Die Theorie der isoelektrischen Fokussierung wurde von Svensson bereits 1961 eingeführt, aber erst nachdem Vesterberg erfolgreich Trägerampholyten zur Erzeugung eines kontinuierlichen pH-Gradienten synthetisiert hatte, war die Technik der isoelektrischen Fokussierung geboren. 1975 kombinierte O'Farrell die isoelektrische Fokussierung mit SDS-PAGE; die sogenannte 2D-Elektrophorese (oder "Protein-Mapping", da jedes Protein aufgrund seiner intrinsischen Ladung und Masse durch eine bestimmte Position gekennzeichnet ist) wurde zu einem wichtigen Werkzeug für die Proteinanalyse.
Ein Quantensprung in der Proteindetektion in Acrylamidgelen (insbesondere 2D-Elektrophorese) war die Einführung von Silberfärbetechniken durch Merrill et al. 1979. Im Vergleich zur bisher vorherrschenden Coomassie®-Färbung wurde die Empfindlichkeit von Mikrogrammen bis Nanogramm stark erhöht. Im selben Jahr führten Towbin et al. den ersten Western Blot durch; er übertrug SDS-PAGE-separierte Proteine auf eine Nitrozellulosemembran.
SDS-PAGE: Gibt es etwas nach Laemmli?
Die von Laemmli geprägte Leistung von SDS-PAGE ist bis dato vorhanden, so dass wir uns fragen müssen, ob es keine Alternativen gibt. Ist dies das Ende der Evolution in der SDS-PAGE? Keine weiteren Techniken oder Verbesserungen in den letzten 40 Jahren? - Natürlich gibt es welche!
Es gibt einige Klassen von Proteinen, die sich in SDS-PAGE anomal verhalten: Glykoproteine, stark basische Proteine (positiv geladen) und einige hydrophobe Transmembranproteine. Für die hochhydrophilen Glykoproteine bietet der Einsatz von alkalischem Tris-borat-EDTA-Puffer (Poduslo 1981) eine Lösung. Zur Abtrennung von Histonen entwickelten Panyim & Chalkley 1969 saure Harnstoff-Polyacrylamidgele (AU-Gele). Eine Alternative ist das TAU-Gel, das zusätzlich das nichtionische Reinigungsmittel Triton® enthält. TAU-Gele eignen sich zur Identifizierung von Modifikationen von Proteinen wie Acetylierung und Phosphorylierung. Andere hochgeladene Proteine können je nach Größe mit dem kationischen Waschmittel Cetyltrimethylammoniumbromid anstelle von SDS abgetrennt werden (Eley et al. 1979).
Für eine verbesserte Trennung von kleinen Peptiden verwendeten Schägger & Jagow 1987 ein alternatives Tris-Tricine-Puffersystem für SDS-PAGE. Vier Jahre später führten die gleichen Forscher ein diskontinuierliches elektrophoretisches System zur Isolierung von Membranproteinen aus Acrylamidgelen ein. In ihrer blauen nativen Elektrophorese wird Coomassie anstelle von SDS verwendet, um eine Ladungsverlagerung auf den Proteinen zu induzieren. Darüber hinaus dienen Aminocapronsäure und nichtionische Reinigungsmittel wie Triton® X-100 dazu, die Löslichkeit der Membranproteine zu verbessern. Die so entstandenen Farbstoff-Waschmittel-Protein-Komplexe werden innerhalb einer aminocaprolsäurehaltigen PAG getrennt und für Studien zur quartären Struktur durch die zweite Dimension Tricine-SDS-PAGE in die einzelnen Polypeptide aufgelöst. Für kleine Membranproteine wird vorgeschlagen, Coomassie® durch Taurodeoxycholat zu ersetzen.
Die Schönheit der Simplifizierung - Ahn "Single Gel".
Im Jahr 2001 führten Ahn et al. eine vereinfachte Variante des SDS-PAGE-Verfahrens ein, die aus nur einem "einzelnen Gel" mit erhöhter Länge für das Trenngel besteht. In Laemmli's Tris-Glycin-Gelen dient Glycin als langsam laufendes Ion. Im Gegensatz dazu verwenden Ahn Single Gele zu diesem Zweck drei Aminosäuren, nämlich Glycin, Serin und Asparagin. Im ursprünglichen Protokoll von Laemmli arbeitet das Trenngel bei pH-Wert 8,8, während das Einzelgel von Ahn bei pH 7,4 arbeitet. Bei einem solchen mild alkalischen pH-Wert wird die Hydrolyse von Acrylamid minimiert. Es verbessert die Stabilität und Haltbarkeit von Acrylamidlösungen und Gelen. Ahn Gele enthalten kein Reinigungsmittel, aber SDS wird dem laufenden Puffer zugesetzt, um Denaturierungsbedingungen während der Elektrophorese zu ermöglichen. Im Originalpapier von Ahn et al. ist kein Vergleich mit anderen SDS-PAGE-Varianten dargestellt. Wir wollten die Unterschiede in der Performance von Gelen nach Ahn oder Laemmli untersuchen. Zu diesem Zweck haben wir die Gelelektrophorese an 10 % Polyacrylamidgelen nach beiden Protokollen durchgeführt. Zusätzlich untersuchten wir den Transfer von Proteinen auf Blotting-Membranen in einem Western Blot Assay. Unsere Ergebnisse in Kürze:
1. Elektrophorese.
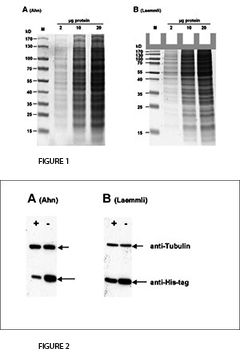
Der erste deutliche Unterschied zwischen Laemmli und Ahn-Gelen ist die Länge des Trenngels für die gleiche Art von Gelkassette. Einzelgele nach Ahn ergeben längere Trenngele (Abb. 1, SDS-PAGE des Gesamtproteins aus HEK293T Zelllysat mit 10 % Acrylamidgelen nach Ahn et al. (A) und Laemmli (B). Coomassie® gefärbte Gele zeigen etwa 20 prominente Proteinbänder mit unterschiedlichen Größenverteilungen für beide Geltypen. Die Migrationsmuster der Proteine waren für beide Geltypen unterschiedlich. Proteine mit einer Molekularmasse von mehr als 70 kD erscheinen auf Laemmli Tris-Glycin-Gelen komprimiert, während Markerproteine mit einem Ahn Single-Gel linear über einen weiten Größenbereich verteilt wurden...). Die Migrationsmuster für Proteine unterscheiden sich jedoch zwischen Laemmli Tris-gylcine und Ahn Single-Gelen in verschiedenen Molekulargewichtsbereichen. Für Proteine von 15 - 70 Kilo Dalton (kD) zeigen 10 % Laemmli Gele die höchste Auflösung, während 10 % Ahn Gele ein lineares Migrationsmuster für Proteingrößen von 25 - 130 kD zeigen. Beim Versuch, eine Vielzahl von Proteingrößen auf demselben Gel zu trennen, gibt es zwei Möglichkeiten: Ahn Einzelgele scheinen sich besonders gut für mittel- und hochmolekulare Proteine zu eignen. Laemmli Tris-Glycin-Gele eignen sich am besten für kleine und mittelgroße Proteine bei gleicher Acrylamidpolymer-Konzentration.
2. Stabilität.
We also studied the stability of acrylamide solutions according to Ahn et al. by storing them for a period of up to six months at 2-8°C. Polyacrylamide gels made from such solutions did not show any significant effect on quality even after prolonged storage. However, cast gels should be used within a few days since the pH value within the gel changes rapidly. This holds true for Laemmli Tris-glycine gels as well as for Ahn single gels.
Wir haben auch die Stabilität von Acrylamidlösungen nach Ahn et al. untersucht, indem wir sie für einen Zeitraum von bis zu sechs Monaten bei 2-8°C gelagert haben. Aus solchen Lösungen hergestellte Polyacrylamidgele zeigten auch nach längerer Lagerung keinen signifikanten Einfluss auf die Qualität. Allerdings sollten gegossene Gele innerhalb weniger Tage verwendet werden, da sich der pH-Wert im Gel schnell ändert. Dies gilt sowohl für Laemmli Tris-Glycin-Gele als auch für Ahn-Einzelgele.
3. Western Blotting.
Für den Transfer von Proteinen variiert die Leistung je nach Molekularmasse der Proteine (Abb. 2, Western Blot of Total Protein aus HEK293T-Zellen nach SDS-PAGE nach Ahn et al. (A) und Laemmli (B). Zwei verschiedene Proteine wurden mit spezifischen Antikörpern nachgewiesen: Tubulin und ein 25 kD his-tagged Protein. Die Expression von Tubulin war in allen Zellen gleich. Der zelluläre (oder proteasomale) Proteinabbau des his-tagged Proteins wurde in den Experimenten induziert (+) oder nicht induziert (-). Tubulin wurde effizienter von Ahn-Gelen auf PVDF-Membranen übertragen als von Laemmli Tris-Glycin-Gelen, während die Übertragung des 25 kD his-tagged Proteins weniger effizient war als bei gleichwertigen Ahn-Einzelgelen...). Tubulin, ein Protein von 66 kD, wurde aus Laemmli-Gelen 55 ± 4 % weniger effizient übertragen als Ahn-Einzelgele. Im Gegensatz dazu war die Übertragung eines 25 kD his-tag-Proteins aus Ahn-Gelen um 32 ± 12 % (n=4) weniger effizient. Insgesamt ist der Transfer von kleinen Proteinen mit den Tris-Glycin-Gelen von Laemmli besser, während Ahn-Einzelgele für Proteine mit höherer Molekularmasse besser abschneiden.
4. Handhabung.
Den befragten Anwendern gefiel das schnelle und komfortable Verfahren nach Ahn et al. Das Protokoll wird von Labors, die bereits das "klassische" Laemmli-Protokoll verwenden, leicht übernommen, da alle Geräte, Puffer und Lösungen für beide SDS-PAGE-Verfahren gleich sind. Andere, skeptischere Autoren bewerteten auch Ahn's Single-Gel-System und bestätigten schließlich die gute Leistung. Laut G. Fritz (von der Universität Zürich, in Rehm 2007) sind zusätzliche Vorteile von Ahn-Gelen: 1. sie laufen recht schön (neigen nicht dazu, "Smilies" zu produzieren), 2. Gele arbeiten mit höheren Polyacrylamidkonzentrationen (bis zu 18 %), 3. sie vermeiden das schleimige Sammelgel.
Perspektiven
In der Forschung werden ständig neue Methoden entwickelt, die neue Erkenntnisse schaffen. Einige der Methoden, wie z.B. die Gelelektrophorese, überstehen den Test der Zeit. Klassische Methoden wurden verbessert, angepasst und im Laufe der Jahre um neue Funktionen erweitert. ITW Reagents ist stets bestrebt, neue oder verbesserte Anwendungsprotokolle zu entwickeln, um vielversprechende Produkte mit den Kunden zu teilen.
Literatur
- Ahn, T. et al. (2001) Anal. Biochem. 291, 300-303
- Davies, B.J. (1964) Ann. N. Y. Acad. Sci. 121, 404-427
- Eley et al. (1979) Anal. Biochem. 92, 411-419
- Fazekas de St. Groth, S. et al. (1963) Biochim. Biophys. Acta 71, 377-391
- Gordon, A.H. et al. (1950) Coll. Czech. Chem. Com. 15, 1
- Grabar, P. & Williams, C.A. (1953) Biochim. Biophys. Acta 10, 193-194
- Hjertén, S. (1961) Biochim. Biophys. Acta 53, 514-517
- Hjertén, S. (1967) Chromatogr. Rev. 9, 122-219
- Hjertén, S. (1983) J. Chromatogr. 270, 1-6
- Jorgenson, J.W. & Lukacs, K.D. (1981) Anal. Chem. 53, 1298
- Kendall, J. & Crittenden, F.D. (1923) Proc. Natl. Acad. Sci. USA 9, 75
- von Klobusitzky, D. & König, P. (1939) Arch. Exptl. Pathol. Pharmakol. Naunyn-Schmiedeberg 192, 271
- Kohlrausch, F. (1897) Wiedemanns Ann. (Ann. Phys. Chem.) 62, 209
- Kohn, J. (1957) Nature 180, 986
- Laemmli, U.K. (1970) Nature 227, 680-685
- Martin, A.J.P. (1942) unpublished results
- Merrill, R.C. et al. (1979) Proc. Natl. Acad. Sci. USA 76, 4335
- O’Farrell, P.H. (1975) J. Biol. Chem. 250, 4007-4021
- Ornstein, L. (1964) Ann. N. Y. Acad. Sci. 121, 321-349
- Panyim, S. & Chalkley, R. (1969) Arch. Biochem. Biophys. 130, 337-346
- Poduslo, J.F. (1981) Anal. Biochem. 114, 131
- Raymond, S. & Weintraub, L. (1959) Science 130, 711
- Reuss, F.F. (1809) Memoires de la Société Imperiale de Naturalistes de Moskou 2, 327
- Sanger, F. & Coulson, A.R. (1975) J. Mol. Biol. 94, 441-448
- Schägger, H. & von Jagow, G. (1987) Anal. Biochem. 166, 368-379
- Schägger, H. & von Jagow, G. (1991) Anal. Biochem. 199, 223-231
- Schwartz, D.C. & Cantor, C.R. (1984) Cell 37, 67-75
- Shapiro, A.L. et al. (1967) Biochim. Biophys. Res. Commun. 28, 815-820
- Smithies, O. (1955) Biochem. J. 61, 629-641
- Svensson, H. (1961) Acta Chem. Scand. 15, 325-341
- Tiselius, A. (1930) Nova Acta Regiae Societatis Scientarum Upsaliensis Ser IV, Vol. 7, No. 4
- Tiselius, A. (1937) Trans. Faraday Soc. 33, 524-531
- Towbin et al. (1979) Proc. Natl. Acad. Sci. USA 76, 4350–4354
- Vesterberg, O. (1969) Acta Chem. Scand. 23, 2653
- Weber, K & Osborn, M. (1969) J. Biol. Chem. 244, 4406
Reviews: - Everaerts, F.M., Becker, J.L., & Verheggen, T.P.E.M. (1976) “Isotachophoresis: Theory, Instrumentation, and Applications”; Elsevier, Amsterdam
- Michov, B. (1995) „Elektrophorese: Theorie und Praxis“; Walter de Gruyter & Co, Berlin
- Vesterberg, O. (1993) “A short history of electrophoretic methods”; Electrophoresis, 14, 1243-1249
- Righetti, P.G. (2005) “Electrophoresis: The march of pennies, the march of dimes”; Journal of Chromatography A, 1079, 24-40
- Westermeier, R. (2004) “Electrophoresis in Practice: A Guide to Methods and Applications of DNA and Protein Separations”; 4th ed. Wiley-VCH, Weinheim